Mutations in the FUS gene cause aggressive amyotrophic lateral sclerosis (ALS-FUS). Beyond mRNA, FUS generates partially processed transcripts retaining introns 6 and 7. We demonstrate that these FUSint6&7-RNA molecules form nuclear condensates, scaffolded by the highly structured intron 7 and associated with nuclear speckles. Using hybridization-proximity labeling proteomics, we show that the FUSint6&7-RNA condensates are enriched for splicing factors and the N6-methyladenosine (m6A) reader YTHDC1. These ribonucleoprotein structures facilitate posttranscriptional FUS splicing and depend on m6A/YTHDC1 for integrity. In cells expressing mutant FUS, FUSint6&7-RNAs become hypermethylated, which in turn stimulates their condensation and splicing. We further show that FUS protein is repelled by m6A. Thus, ALS-FUS mutations may cause abnormal activation of FUS posttranscriptional splicing through altered RNA methylation. Notably, ectopic expression of FUS intron 7 sequences dissolves endogenous FUSint6&7-RNA condensates, down-regulating FUS mRNA and protein. Our findings reveal a condensation-dependent mechanism regulating FUS splicing, with possible therapeutic implications for ALS.
Product Citations: 21
In Science Advances on 25 July 2025 by Huang, W. P., Kumar, V., et al.
Single Antisense Oligonucleotides Correct Diverse Splicing Mutations in Hotspot Exons.
In Proceedings of the National Academy of Sciences of the United States of America on 24 June 2025 by Duan, C., Rong, S., et al.
Mutations that impact splicing play a significant role in disease etiology but are not fully understood. To characterize the impact of exonic variants on splicing in 71 clinically actionable disease genes in asymptomatic people, we analyzed 32,112 exonic mutations from ClinVar and Geisinger MyCode using a minigene reporter assay. We identify 1,733 splice-disrupting mutations, with the most extreme variants likely being deleterious. We report that these variants are not distributed evenly across exons but are mostly concentrated in the ~8% of exons that are most susceptible to splicing mutations (i.e., hotspot exons). We demonstrate how multiple, splice-disrupting mutations in these exons can be reverted by the same ASOs targeting the splice sites of either their upstream or downstream flanking exons. This finding supports the feasibility of developing single therapeutic ASOs that could revert all splice-altering variants localized to a particular exon.
Covalent targeting of splicing in T cells.
In Cell Chemical Biology on 16 January 2025 by Scott, K. A., Kojima, H., et al.
Despite significant interest in therapeutic targeting of splicing, few chemical probes are available for the proteins involved in splicing. Here, we show that elaborated stereoisomeric acrylamide EV96 and its analogues lead to a selective T cell state-dependent loss of interleukin 2-inducible T cell kinase (ITK) by targeting one of the core splicing factors SF3B1. Mechanistic investigations suggest that the state-dependency stems from a combination of differential protein turnover rates and extensive ITK mRNA alternative splicing. We further introduce the most comprehensive list to date of proteins involved in splicing and leverage cysteine- and protein-directed activity-based protein profiling with electrophilic scout fragments to demonstrate covalent ligandability for many classes of splicing factors and splicing regulators in T cells. Taken together, our findings show how chemical perturbation of splicing can lead to immune state-dependent changes in protein expression and provide evidence for the broad potential to target splicing factors with covalent chemistry.
Published by Elsevier Ltd.
One-Size-Fits-Many: Antisense oligonucleotides for rescuing splicing mutations in hotspot exons
Preprint on BioRxiv : the Preprint Server for Biology on 8 December 2024 by Duan, C., Rong, S., et al.
ABSTRACT Mutations that impact splicing play a significant role in disease etiology but are not fully understood. To characterize the impact of exonic variants on splicing in 71 clinically-actionable disease genes in asymptomatic people, we analyzed 32,112 exonic mutations from ClinVar and Geisinger MyCode using a minigene reporter assay. We identify 1,733 splice-disrupting mutations, of which the most extreme 1-2% of variants are likely to be deleterious. We report that these variants are not distributed evenly across exons but are mostly concentrated in the ∼8% of exons that are most susceptible to splicing mutations (i.e. hotspot exons). We demonstrate that splicing defects in these exons can be reverted by ASOs targeting the splice sites of either their upstream or downstream flanking exons. This finding supports the feasibility of developing single therapeutic ASOs that could revert all splice-altering variants localized to a particular exon.
In Birth Defects Research on 1 November 2024 by Hoshino, Y., Liu, S., et al.
Mutations in genes encoding spliceosome components result in craniofacial structural defects in humans, referred to as spliceosomopathies. The SF3b complex is a crucial unit of the spliceosome, but model organisms generated through genetic modification of the complex do not perfectly mimic the phenotype of spliceosomopathies. Since the phenotypes are suggested to be determined by the extent of spliceosome dysfunction, an alternative experimental system that can seamlessly control SF3b function is needed.
To establish another experimental system for model organisms elucidating relationship between spliceosome function and human diseases, we administered Pladienolide-B (PB), a SF3b complex inhibitor, to mouse and zebrafish embryos and assessed resulting phenotypes.
PB-treated mouse embryos exhibited neural tube defect and exencephaly, accompanied by apoptosis and reduced cell proliferation in the neural tube, but normal structure in the midface and jaw. PB administration to heterozygous knockout mice of Sf3b4, a gene coding for a SF3b component, influenced the formation of cranial neural crest cells (CNCCs). Despite challenges in continuous PB administration and a high death rate in mice, PB was stably administered to zebrafish embryos, resulting in prolonged survival. Brain, cranial nerve, retina, midface, and jaw development were affected, mimicking spliceosomopathy phenotypes. Additionally, alterations in cell proliferation, cell death, and migration of CNCCs were detected.
We demonstrated that zebrafish treated with PB exhibited phenotypes similar to those observed in human spliceosomopathies. This experimental system may serve as a valuable research tool for understanding spliceosome function and human diseases.
© 2024 The Author(s). Birth Defects Research published by Wiley Periodicals LLC.
In Aging Cell on 1 November 2024 by Deschênes, M., Durand, M., et al.
Fig.3.E
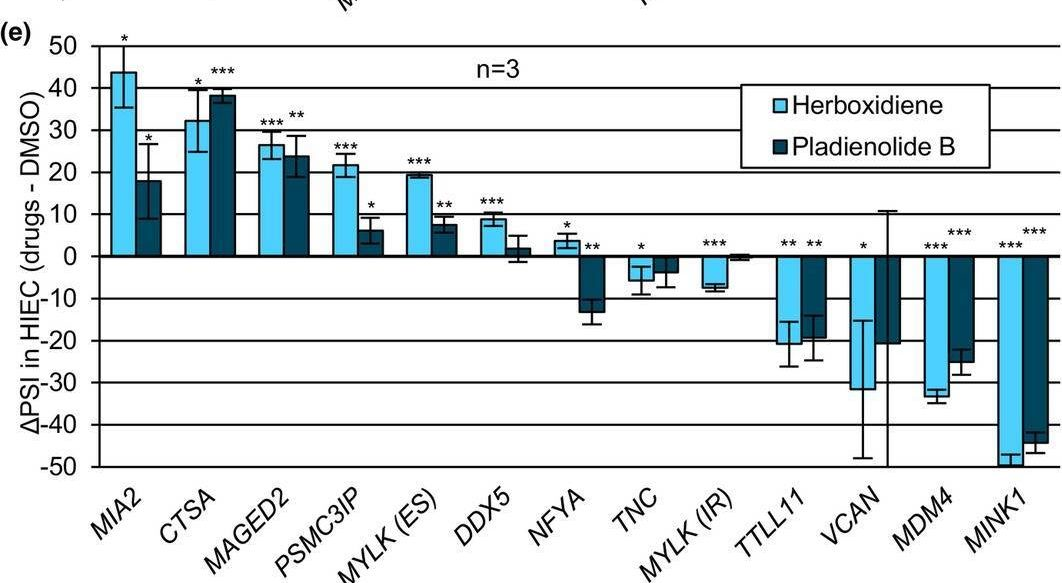
Collected and cropped from Aging Cell by CiteAb, provided under a CC-BY license
Image 1 of 2
In Aging Cell on 1 November 2024 by Deschênes, M., Durand, M., et al.
Fig.3.D
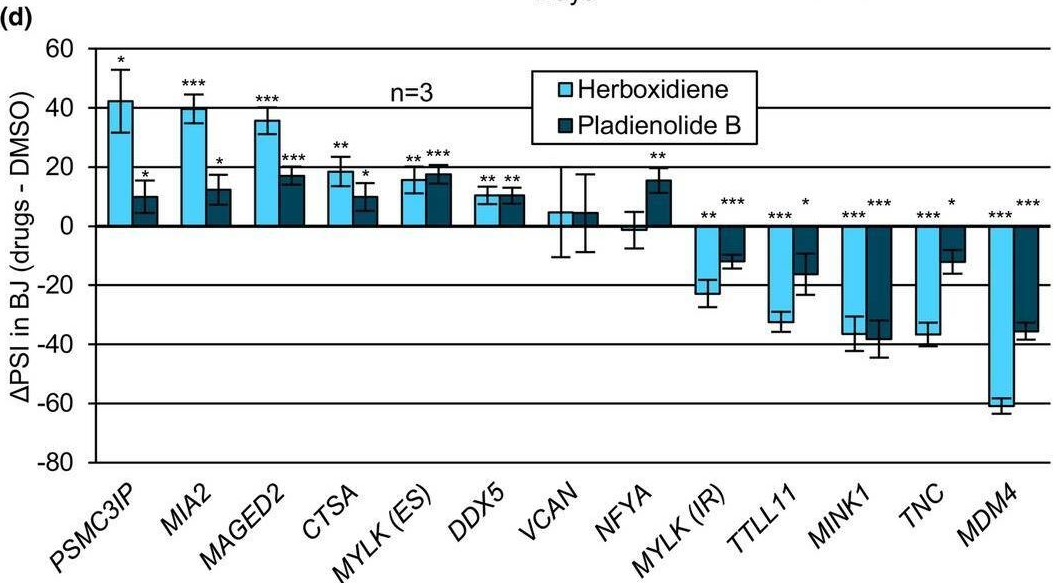
Collected and cropped from Aging Cell by CiteAb, provided under a CC-BY license
Image 1 of 2