Overexpression of the myristolated alanine-rich C kinase substrate (MARCKS) occurs in vascular proliferative diseases such as restenosis after bypass surgery. MARCKS knockdown results in arrest of vascular smooth muscle cell (VSMC) proliferation with little effect on endothelial cell (EC) proliferation. We sought to identify the mechanism of differential regulation by MARCKS of VSMC and EC proliferation in vitro and in vivo.siRNA-mediated MARCKS knockdown in VSMCs inhibited proliferation and prevented progression from phase G0/G1 to S. Protein expression of the cyclin-dependent kinase inhibitor p27kip1, but not p21cip1 was increased by MARCKS knockdown. MARCKS knockdown did not affect proliferation in VSMCs derived from p27kip1-/- mice indicating that the effect of MARCKS is p27kip1-dependent. MARCKS knockdown resulted in decreased phosphorylation of p27kip1 at threonine 187 and serine 10 as well as, kinase interacting with stathmin (KIS), cyclin D1, and Skp2 expression. Phosphorylation of p27kip1 at serine 10 by KIS is required for nuclear export and degradation of p27kip1. MARCKS knockdown caused nuclear trapping of p27kip1. Both p27kip1 nuclear trapping and cell cycle arrest were released by overexpression of KIS, but not catalytically inactive KIS. In ECs, MARCKS knockdown paradoxically increased KIS expression and cell proliferation. MARCKS knockdown in a murine aortic injury model resulted in decreased VSMC proliferation determined by bromodeoxyuridine (BrdU) integration assay, and inhibition of vascular wall thickening. MARCKS knockdown increased the rate of re-endothelialization.MARCKS knockdown arrested VSMC cell cycle by decreasing KIS expression. Decreased KIS expression resulted in nuclear trapping of p27kip1 in VSMCs. MARCKS knockdown paradoxically increased KIS expression in ECs resulting in increased EC proliferation. MARCKS knockdown significantly attenuated the VSMC proliferative response to vascular injury, but accelerated reestablishment of an intact endothelium. MARCKS is a novel translational target with beneficial cell type-specific effects on both ECs and VSMCs.
Product Citations: 8
In PLoS ONE on 4 November 2015 by Yu, D., Makkar, G., et al.
-
WB
-
Homo sapiens (Human)
BRAF inhibitor-associated ERK activation drives development of chronic lymphocytic leukemia.
In The Journal of Clinical Investigation on 1 November 2014 by Yaktapour, N., Meiss, F., et al.
Patients with BRAFV600E/K-driven melanoma respond to the BRAF inhibitor vemurafenib due to subsequent deactivation of the proliferative RAS/RAF/MEK/ERK pathway. In BRAF WT cells and those with mutations that activate or result in high levels of the BRAF activator RAS, BRAF inhibition can lead to ERK activation, resulting in tumorigenic transformation. We describe a patient with malignant melanoma who developed chronic lymphocytic leukemia (CLL) in the absence of RAS mutations during vemurafenib treatment. BRAF inhibition promoted patient CLL proliferation in culture and in murine xenografts and activated MEK/ERK in primary CLL cells from additional patients. BRAF inhibitor-driven ERK activity and CLL proliferation required B cell antigen receptor (BCR) activation, as inhibition of the BCR-proximal spleen tyrosine kinase (SYK) reversed ERK hyperactivation and proliferation of CLL cells from multiple patients, while inhibition of the BCR-distal Bruton tyrosine kinase had no effect. Additionally, the RAS-GTP/RAS ratio in primary CLL cells exposed to vemurafenib was reduced upon SYK inhibition. BRAF inhibition increased mortality and CLL expansion in mice harboring CLL xenografts; however, SYK or MEK inhibition prevented CLL proliferation and increased animal survival. Together, these results suggest that BRAF inhibitors promote B cell malignancies in the absence of obvious mutations in RAS or other receptor tyrosine kinases and provide a rationale for combined BRAF/MEK or BRAF/SYK inhibition.
-
Cancer Research
In British Journal of Haematology on 1 October 2013 by Klitgaard, J. L., Koefoed, K., et al.
The treatment of chronic lymphocytic leukaemia (CLL) has been improved by introduction of monoclonal antibodies (mAbs) that exert their effect through secondary effector mechanisms. CLL cells are characterized by expression of CD5 and CD23 along with CD19 and CD20, hence anti-CD5 Abs that engage secondary effector functions represent an attractive opportunity for CLL treatment. Here, a repertoire of mAbs against human CD5 was generated and tested for ability to induce complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) both as single mAbs and combinations of two mAbs against non-overlapping epitopes on human CD5. The results demonstrated that combinations of two mAbs significantly increased the level of CDC compared to the single mAbs, while no enhancement of ADCC was seen with anti-CD5 mAb combinations. High levels of CDC and ADCC correlated with low levels of Ab-induced CD5 internalization and degradation. Importantly, an anti-CD5 mAb combination enhanced CDC of CLL cells when combined with the anti-CD20 mAbs rituximab and ofatumumab as well as with the anti-CD52 mAb alemtuzumab. These results suggest that an anti-CD5 mAb combination inducing CDC and ADCC may be effective alone, in combination with mAbs against other targets or combined with chemotherapy for CLL and other CD5-expressing haematological or lymphoid malignancies.
© 2013 John Wiley & Sons Ltd.
-
Cardiovascular biology
In Cytometry. Part B, Clinical Cytometry on 1 May 2008 by Bahler, D. W., Hartung, L., et al.
Enumeration of neoplastic T-cells in peripheral blood specimens is necessary for the diagnosis of Sézary syndrome (SS) and monitoring treatment responses. Because neoplastic T-cells in SS can be difficult to identify by morphology alone, flow cytometry immunophenotyping is often utilized. However, the reported immunophenotypic criteria for identifying neoplastic T-cells in SS are variable, not present in all cases, or sometimes found in reactive T-cell populations. Peripheral blood lymphocytes from 33 cases of SS were evaluated for the expression of pan-T cell antigens and killer cell immunoglobulin-like MHC receptors (KIR) CD158a, CD158b, CD158e, CD158i, and CD158k by multiparameter flow cytometry using monoclonal antibodies EB6, GL183, FES172, Z27, and Q66. A variety of abnormalities related to expression of pan-T-cell antigens typical of neoplastic T-cells were observed. Expression of CD158k was observed in 32/33 cases and restricted to the phenotypically abnormal T-cell populations, while expression of other KIR was mostly negative. Our findings confirm and extend recent reports by one group that CD158k is expressed by most SS cases. Moreover, our observation that CD4 positive, CD7 negative T-cells are mostly CD158k negative further suggests that CD158k may be able to help identify and enumerate neoplastic T-cells in SS even when present at low levels.
-
Immunology and Microbiology
In The Journal of Cell Biology on 18 August 2003 by Lin, J. & Weiss, A.
CD148 is a receptor-like protein tyrosine phosphatase up-regulated on T cells after T cell receptor (TCR) stimulation. To examine the physiologic role of CD148 in TCR signaling, we used an inducible CD148-expressing Jurkat T cell clone. Expression of CD148 inhibits NFAT (nuclear factor of activated T cells) activation induced by soluble anti-TCR antibody, but not by antigen-presenting cells (APCs) loaded with staphylococcal enterotoxin superantigen (SAg) or immobilized anti-TCR antibody. Immunofluorescence microscopy revealed that the extracellular domain of CD148 mediates its exclusion from the immunologic synapse, sequestering it from potential substrates. Targeting of the CD148 phosphatase domain to the immunologic synapse potently inhibited NFAT activation by all means of triggering through the TCR. These data lead us to propose a model where CD148 function is regulated in part by exclusion from substrates in the immunologic synapse. Upon T cell-APC disengagement, CD148 can then access and dephosphorylate substrates to down-regulate prolongation of signaling.
-
Cell Biology
-
Immunology and Microbiology
-
Neuroscience
In PLoS One on 4 November 2015 by Yu, D., Makkar, G., et al.
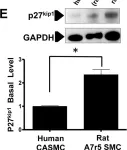
Fig.5.E

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 1