Oncogene activation in normal untransformed cells induces DNA replication stress and creates a dependency on DNA damage response (DDR) mechanisms for cell survival. Different oncogenic stimuli signal via distinct mechanisms in every cancer setting. The DDR is also pathologically reprogrammed and deployed in diverse ways in different cancers. Because mutant KRAS is the driver oncogene in 90% of pancreatic ductal adenocarcinomas (PDACs), here we have investigated DDR mechanisms by which KRAS-induced DNA replication stress is tolerated in normal human pancreatic epithelial cells [human pancreatic nestin-expressing (HPNE) cells]. Using a candidate screening approach, we identify TRIP13 as a KRASG12V-induced messenger RNA that is also expressed at high levels in PDAC relative to normal tissues. Using genetic and pharmacological tools, we show that TRIP13 is necessary to sustain ongoing DNA synthesis and viability specifically in KRASG12V-expressing cells. TRIP13 promotes survival of KRASG12V-expressing HPNE cells in a homologous recombination (HR)-dependent manner. KRASG12V-expressing HPNE cells lacking TRIP13 acquire hallmark HR deficiency phenotypes, including sensitivity to inhibitors of translesion synthesis and poly-ADP ribose polymerase. Established PDAC cell lines are also sensitized to intrinsic DNA damage and therapy-induced genotoxicity following TRIP13 depletion. Taken together, our results expose TRIP13 as an attractive new and therapeutically tractable vulnerability of KRAS-mutant PDAC.
© The Author(s) 2025. Published by Oxford University Press on behalf of NAR Cancer.
Product Citations: 210
In NAR Cancer on 1 March 2025 by Anand, J. R., Droby, G. N., et al.
-
Cancer Research
-
Genetics
TRIP13 protects pancreatic cancer cells against intrinsic and therapy-induced DNA replication stress
Preprint on BioRxiv : the Preprint Server for Biology on 27 January 2025 by Anand, J. R., Droby, G. N., et al.
Oncogene activation in normal untransformed cells induces DNA replication stress and creates a dependency on DNA Damage Response (DDR) mechanisms for cell survival. Different oncogenic stimuli signal via distinct mechanisms in every cancer setting. The DDR is also pathologically re-programmed and deployed in diverse ways in different cancers. Because mutant KRAS is the driver oncogene in 90% of Pancreatic Ductal Adenocarcinomas (PDAC), here we have investigated DDR mechanisms by which KRAS-induced DNA replication stress is tolerated in normal human pancreatic epithelial cells (HPNE). Using a candidate screening approach, we identify TRIP13 as a KRASG12V-induced mRNA that is also expressed at high levels in PDAC relative to normal tissues. Using genetic and pharmacological tools, we show that TRIP13 is necessary to sustain ongoing DNA synthesis and viability specifically in KRASG12V-expressing cells. TRIP13 promotes survival of KRASG12V-expressing HPNE cells in a Homologous Recombination (HR)-dependent manner. KRASG12V-expressing HPNE cells lacking TRIP13 acquire hallmark HR-deficiency (HRD) phenotypes including sensitivity to inhibitors of Trans-Lesion Synthesis (TLS) and Poly-ADP Ribose Polymerase (PARP). Established PDAC cell lines are also sensitized to intrinsic DNA damage and therapy-induced genotoxicity following TRIP13-depletion. Taken together our results expose TRIP13 as an attractive new and therapeutically-tractable vulnerability of KRAS-mutant PDAC.
-
Cancer Research
-
Genetics
In Nucleic Acids Research on 27 November 2024 by Yang, Y., Jayaprakash, D., et al.
RNF168 orchestrates a ubiquitin-dependent DNA damage response to regulate the recruitment of repair factors, such as 53BP1 to DNA double-strand breaks (DSBs). In addition to its canonical functions in DSB signaling, RNF168 may facilitate DNA replication fork progression. However, the precise role of RNF168 in DNA replication remains unclear. Here, we demonstrate that RNF168 is recruited to DNA replication factories in a manner that is independent of the canonical DSB response pathway regulated by Ataxia-Telangiectasia Mutated (ATM) and RNF8. We identify a degenerate Proliferating Cell Nuclear Antigen (PCNA)-interacting peptide (DPIP) motif in the C-terminus of RNF168, which together with its Motif Interacting with Ubiquitin (MIU) domain mediates binding to mono-ubiquitylated PCNA at replication factories. An RNF168 mutant harboring inactivating substitutions in its DPIP box and MIU1 domain (termed RNF168 ΔDPIP/ΔMIU1) is not recruited to sites of DNA synthesis and fails to support ongoing DNA replication. Notably, the PCNA interaction-deficient RNF168 ΔDPIP/ΔMIU1 mutant fully rescues the ability of RNF168-/- cells to form 53BP1 foci in response to DNA DSBs. Therefore, RNF168 functions in DNA replication and DSB signaling are fully separable. Our results define a new mechanism by which RNF168 promotes DNA replication independently of its canonical functions in DSB signaling.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
-
Biochemistry and Molecular biology
-
Genetics
Specific origin selection and excess functional MCM2-7 loading in ORC-deficient cells
Preprint on BioRxiv : the Preprint Server for Biology on 30 October 2024 by Shibata, Y., Peycheva, M., et al.
ABSTRACT The six subunit Origin Recognition Complex (ORC) loads excess MCM2-7 on chromosomes to promote initiation of DNA replication and is believed to be important for origin specification. Mapping of origins in cancer cell lines engineered to delete three of the subunits, ORC1 , ORC2 or ORC5 shows that specific origins are still used and are mostly at the same sites in the genome as in wild type cells. The few hundred origins that were up-regulated in the absence of ORC suggest that GC/TA skewness and simple repeat sequences facilitate, but are not essential for, origin selection in the absence of the six-subunit ORC. Despite the lack of ORC, excess MCM2-7 is still loaded at comparable rates in G1 phase to license reserve origins and is also repeatedly loaded in the same S phase to permit re-replication. Thus, origin specification and excess MCM2-7 loading on origins do not require the six-subunit ORC in human cancer cell lines.
-
Homo sapiens (Human)
In Cellular Oncology (Dordrecht) on 1 June 2024 by Cokelaere, C., Dok, R., et al.
TIPRL1 (target of rapamycin signaling pathway regulator-like 1) is a known interactor and inhibitor of protein phosphatases PP2A, PP4 and PP6 - all pleiotropic modulators of the DNA Damage Response (DDR). Here, we investigated the role of TIPRL1 in the radiotherapy (RT) response of Head and Neck Squamous Cell Carcinoma (HNSCC).
TIPRL1 mRNA (cBioportal) and protein expression (immunohistochemistry) in HNSCC samples were linked with clinical patient data. TIPRL1-depleted HNSCC cells were generated by CRISPR/Cas9 editing, and effects on colony growth, micronuclei formation (microscopy), cell cycle (flow cytometry), DDR signaling (immunoblots) and proteome (mass spectrometry) following RT were assessed. Mass spectrometry was used for TIPRL1 phosphorylation and interactomics analysis in irradiated cells.
TIPRL1 expression was increased in tumor versus non-tumor tissue, with high tumoral TIPRL1 expression associating with lower locoregional control and decreased survival of RT-treated patients. TIPRL1 deletion in HNSCC cells resulted in increased RT sensitivity, a faster but prolonged cell cycle arrest, increased micronuclei formation and an altered proteome-wide DDR. Upon irradiation, ATM phosphorylates TIPRL1 at Ser265. A non-phospho Ser265Ala mutant could not rescue the increased radiosensitivity phenotype of TIPRL1-depleted cells. While binding to PP2A-like phosphatases was confirmed, DNA-dependent protein kinase (DNA-PKcs), RAD51 recombinase and nucleosomal histones were identified as novel TIPRL1 interactors. Histone binding, although stimulated by RT, was adversely affected by TIPRL1 Ser265 phosphorylation.
Our findings underscore a clinically relevant role for TIPRL1 and its ATM-dependent phosphorylation in RT resistance through modulation of the DDR, highlighting its potential as a new HNSCC predictive marker and therapeutic target.
© 2023. Springer Nature Switzerland AG.
-
FC/FACS
-
Homo sapiens (Human)
-
Cancer Research
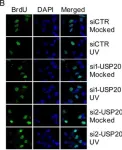
In Nucleic Acids Res on 1 December 2014 by Zhu, M., Zhao, H., et al.
Fig.5.B

-
ICC-IF
-
Collected and cropped from Nucleic Acids Res by CiteAb, provided under a CC-BY license
Image 1 of 3
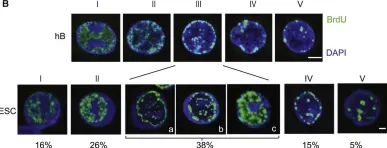
In Cell on 14 February 2013 by Tsubouchi, T., Soza-Ried, J., et al.
Fig.1.B

-
ICC-IF
-
Collected and cropped from Cell by CiteAb, provided under a CC-BY license
Image 1 of 3
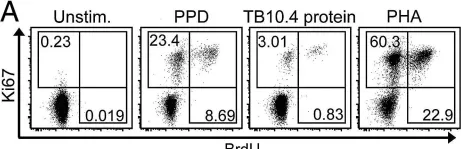
In J Immunol Methods on 31 October 2010 by Soares, A., Govender, L., et al.
Fig.2.A

-
FC/FACS
-
Collected and cropped from J Immunol Methods by CiteAb, provided under a CC-BY license
Image 1 of 3