Rapid eye movement (REM) sleep deprivation triggers mania and induces cardiac fibrosis. Beyond neuroprotection, lithium has cardioprotective potential and antifibrotic activity. This study investigated whether lithium improved REM sleep deprivation-induced cardiac dysfunction and evaluated the potential mechanisms. Transthoracic echocardiography, histopathological analysis, and Western blot analysis were performed in control and REM sleep-deprived rats with or without lithium treatment (LiCl of 1 mmol/kg/day administered by oral gavage for 4 weeks) in vivo and in isolated ventricular preparations. The results revealed that REM sleep-deprived rats exhibited impaired contractility and greater fibrosis than control and lithium-treated REM sleep-deprived rats. Western blot analysis showed that REM sleep-deprived hearts had higher expression levels of transforming growth factor beta (TGF-β), phosphorylated Smad 2/3, and alpha-smooth muscle actin than lithium-treated REM sleep-deprived and control hearts. Moreover, lithium-treated REM sleep-deprived hearts had lower expression of angiotensin II type 1 receptor, phosphorylated nuclear factor kappa B p65, calcium release-activated calcium channel protein 1, transient receptor potential canonical (TRPC) 1, and TRPC3 than REM sleep-deprived hearts. The findings suggest that lithium attenuates REM sleep deprivation-induced cardiac fibrogenesis and dysfunction possibly through the downregulation of TGF-β, angiotensin II, and Ca2+ signaling.
Product Citations: 15
Applications
Reactivity
Research Area
Lithium Treatment Improves Cardiac Dysfunction in Rats Deprived of Rapid Eye Movement Sleep.
In International Journal of Molecular Sciences on 23 September 2022 by Chen, P. H., Chung, C. C., et al.
-
WB
-
Cardiovascular biology
In International Journal of Molecular Sciences on 23 December 2021 by Lee, T. W., Chung, C. C., et al.
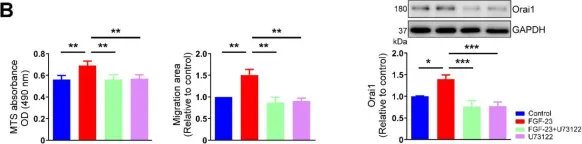
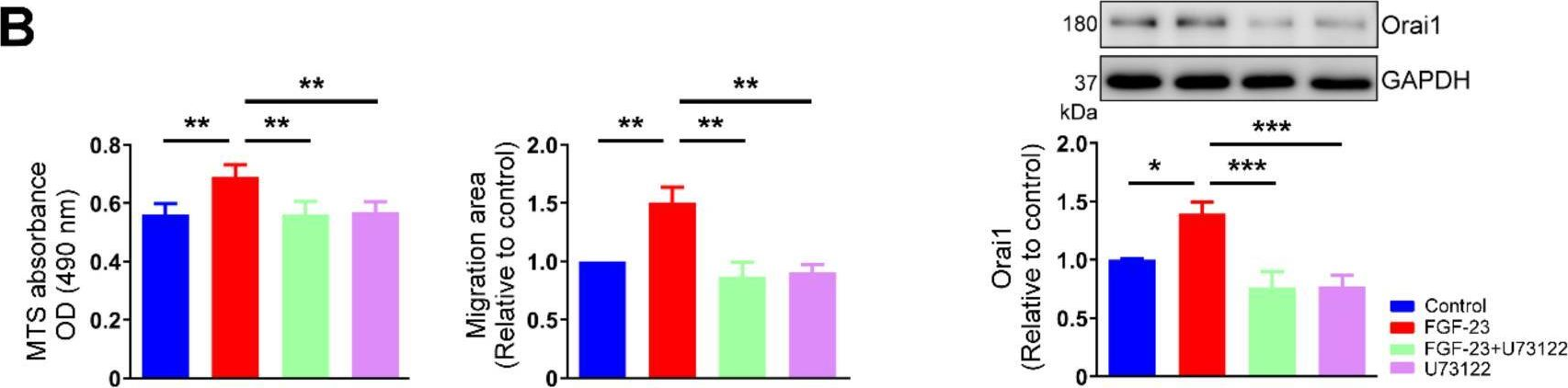
Fibroblast growth factor (FGF)-23 induces hypertrophy and calcium (Ca2+) dysregulation in cardiomyocytes, leading to cardiac arrhythmia and heart failure. However, knowledge regarding the effects of FGF-23 on cardiac fibrogenesis remains limited. This study investigated whether FGF-23 modulates cardiac fibroblast activity and explored its underlying mechanisms. We performed MTS analysis, 5-ethynyl-2'-deoxyuridine assay, and wound-healing assay in cultured human atrial fibroblasts without and with FGF-23 (1, 5 and 25 ng/mL for 48 h) to analyze cell proliferation and migration. We found that FGF-23 (25 ng/mL, but not 1 or 5 ng/mL) increased proliferative and migratory abilities of human atrial fibroblasts. Compared to control cells, FGF-23 (25 ng/mL)-treated fibroblasts had a significantly higher Ca2+ entry and intracellular inositol 1,4,5-trisphosphate (IP3) level (assessed by fura-2 ratiometric Ca2+ imaging and enzyme-linked immunosorbent assay). Western blot analysis showed that FGF-23 (25 ng/mL)-treated cardiac fibroblasts had higher expression levels of calcium release-activated calcium channel protein 1 (Orai1) and transient receptor potential canonical (TRPC) 1 channel, but similar expression levels of α-smooth muscle actin, collagen type IA1, collagen type Ⅲ, stromal interaction molecule 1, TRPC 3, TRPC6 and phosphorylated-calcium/calmodulin-dependent protein kinase II when compared with control fibroblasts. In the presence of ethylene glycol tetra-acetic acid (a free Ca2+ chelator, 1 mM) or U73122 (an inhibitor of phospholipase C, 1 μM), control and FGF-23-treated fibroblasts exhibited similar proliferative and migratory abilities. Moreover, polymerase chain reaction analysis revealed that atrial fibroblasts abundantly expressed FGF receptor 1 but lacked expressions of FGF receptors 2-4. FGF-23 significantly increased the phosphorylation of FGF receptor 1. Treatment with PD166866 (an antagonist of FGF receptor 1, 1 μM) attenuated the effects of FGF-23 on cardiac fibroblast activity. In conclusion, FGF-23 may activate FGF receptor 1 and subsequently phospholipase C/IP3 signaling pathway, leading to an upregulation of Orai1 and/or TRPC1-mediated Ca2+ entry and thus enhancing human atrial fibroblast activity.
-
WB
-
Homo sapiens (Human)
-
Cardiovascular biology
In International Journal of Molecular Sciences on 15 January 2021 by Chen, P. H., Chung, C. C., et al.
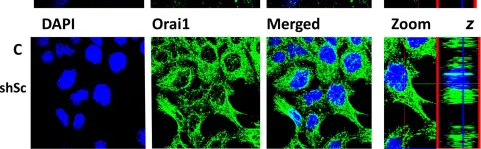
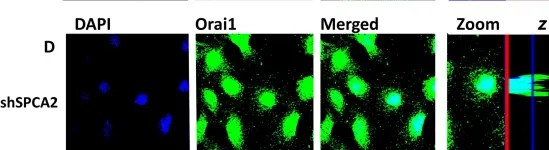
Cardiac fibrosis plays a vital role in the pathogenesis of heart failure. Fibroblast activity is enhanced by increases in store-operated Ca2+ entry (SOCE) and calcium release-activated calcium channel protein 1 (Orai1) levels. Lithium regulates SOCE; however, whether therapeutic concentrations of lithium can be used to inhibit cardiac fibrogenesis is unknown. Migration and proliferation assays, Western blotting, real-time reverse-transcription polymerase chain reaction analysis, and calcium fluorescence imaging were performed in human cardiac fibroblasts treated with or without LiCl at 1.0 mM (i.e., therapeutic peak level) or 0.1 mM (i.e., therapeutic trough level) for 24 h. Results showed that LiCl (0.1 mM, but not 1.0 mM) inhibited the migration and collagen synthesis ability of cardiac fibroblasts. Additionally, thapsigargin-induced SOCE was reduced in fibroblasts treated with LiCl (0.1 mM). The expression level of Orai1 was lower in LiCl (0.1 mM)-treated fibroblasts relative to the fibroblasts without LiCl treatment. Fibroblasts treated with a combination of LiCl (0.1 mM) and 2-APB (10 μM, an Orai1 inhibitor) demonstrated similar migration and collagen synthesis abilities as those in LiCl (0.1 mM)-treated fibroblasts. Altogether, lithium at therapeutic trough levels reduced the migration and collagen synthesis abilities of human cardiac fibroblasts by inhibiting SOCE and Orai1 expression.
-
WB
-
Homo sapiens (Human)
-
Cardiovascular biology
Targeting Orai1-Mediated Store-Operated Ca2+ Entry in Heart Failure.
In Frontiers in Cell and Developmental Biology on 30 October 2020 by Luo, R., Gomez, A. M., et al.
The archetypal store-operated Ca2+ channels (SOCs), Orai1, which are stimulated by the endo/sarcoplasmic reticulum (ER/SR) Ca2+ sensor stromal interaction molecule 1 (STIM1) upon Ca2+ store depletion is traditionally viewed as instrumental for the function of non-excitable cells. In the recent years, expression and function of Orai1 have gained recognition in excitable cardiomyocytes, albeit controversial. Even if its cardiac physiological role in adult is still elusive and needs to be clarified, Orai1 contribution in cardiac diseases such as cardiac hypertrophy and heart failure (HF) is increasingly recognized. The present review surveys our current arising knowledge on the new role of Orai1 channels in the heart and debates on its participation to cardiac hypertrophy and HF.
Copyright © 2020 Luo, Gomez, Benitah and Sabourin.
-
Cardiovascular biology
In Molecular Oncology on 1 January 2020 by Bong, A. H. L., Robitaille, M., et al.
Neuronal calcium sensor-1 (NCS-1) is a positive modulator of IP3 receptors and was recently associated with poorer survival in breast cancers. However, the association between NCS-1 and breast cancer molecular subtypes and the effects of NCS-1 silencing on calcium (Ca2+ ) signaling in breast cancer cells remain unexplored. Herein, we report for the first time an increased expression of NCS-1 in breast cancers of the basal molecular subtype, a subtype associated with poor prognosis. Using MDA-MB-231 basal breast cancer cells expressing the GCaMP6m Ca2+ indicator, we showed that NCS-1 silencing did not result in major changes in cytosolic free Ca2+ increases as a result of endoplasmic reticulum Ca2+ store mobilization. However, NCS-1 silencing suppressed unstimulated basal Ca2+ influx. NCS-1 silencing in MDA-MB-231 cells also promoted necrotic cell death induced by the chemotherapeutic drug doxorubicin (1 µm). The effect of NCS-1 silencing on cell death was phenocopied by silencing of ORAI1, a Ca2+ store-operated Ca2+ channel that maintains Ca2+ levels in the endoplasmic reticulum Ca2+ store and whose expression was significantly positively correlated with NCS-1 in clinical breast cancer samples. This newly identified association between NCS-1 and basal breast cancers, together with the identification of the role of NCS-1 in the regulation of the effects of doxorubicin in MDA-MB-231 breast cancer cells, suggests that NCS-1 and/or pathways regulated by NCS-1 may be important in the treatment of basal breast cancers in women.
© 2019 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
-
WB
-
Homo sapiens (Human)
In Int J Mol Sci on 23 December 2021 by Lee, T. W., Chung, C. C., et al.
Fig.8.B
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
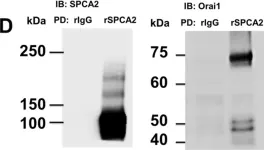
Fig.1.D
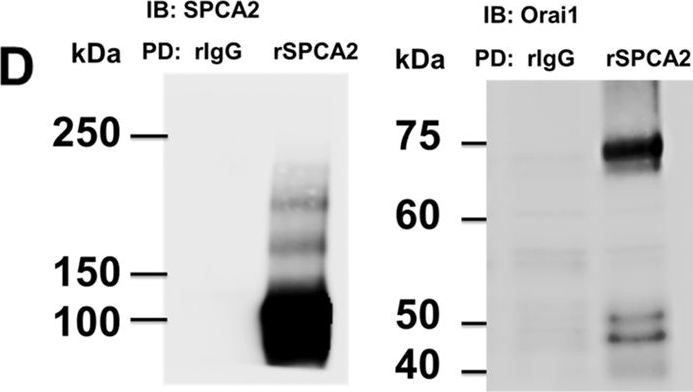
-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
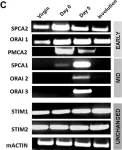
Fig.1.C
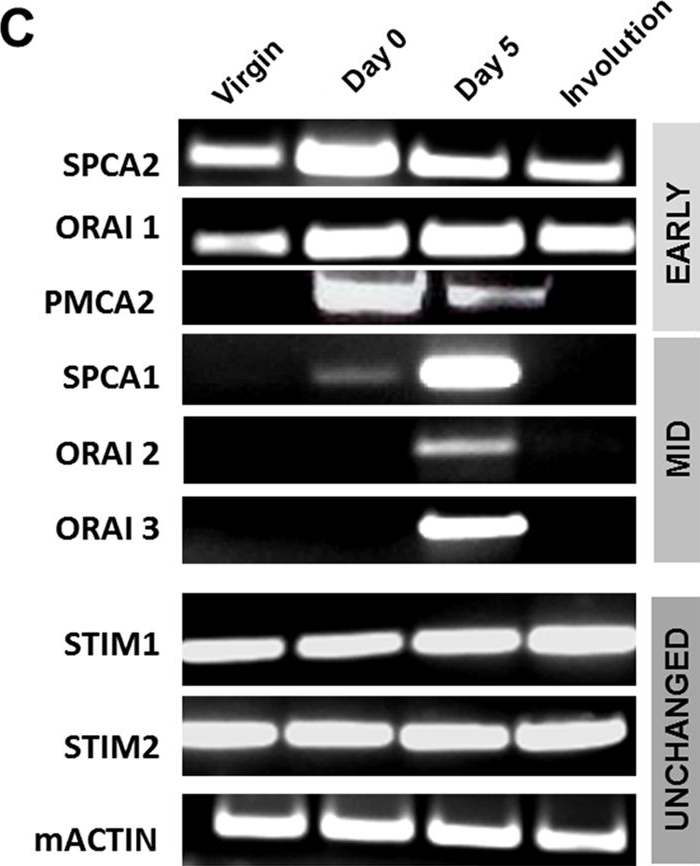
-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
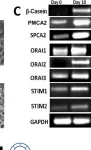
Fig.3.C
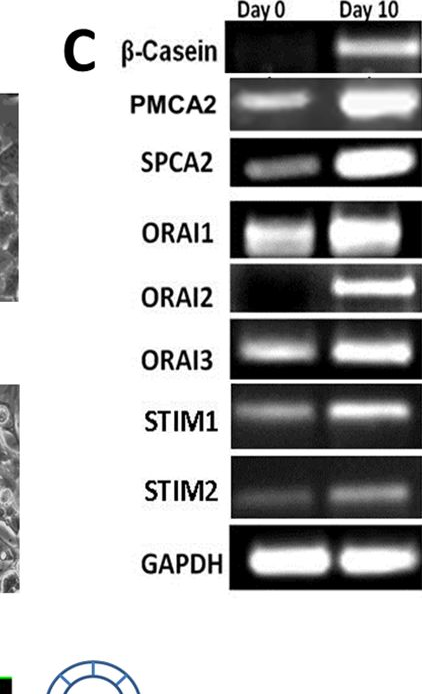
-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
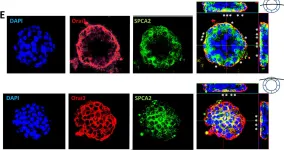
Fig.3.E
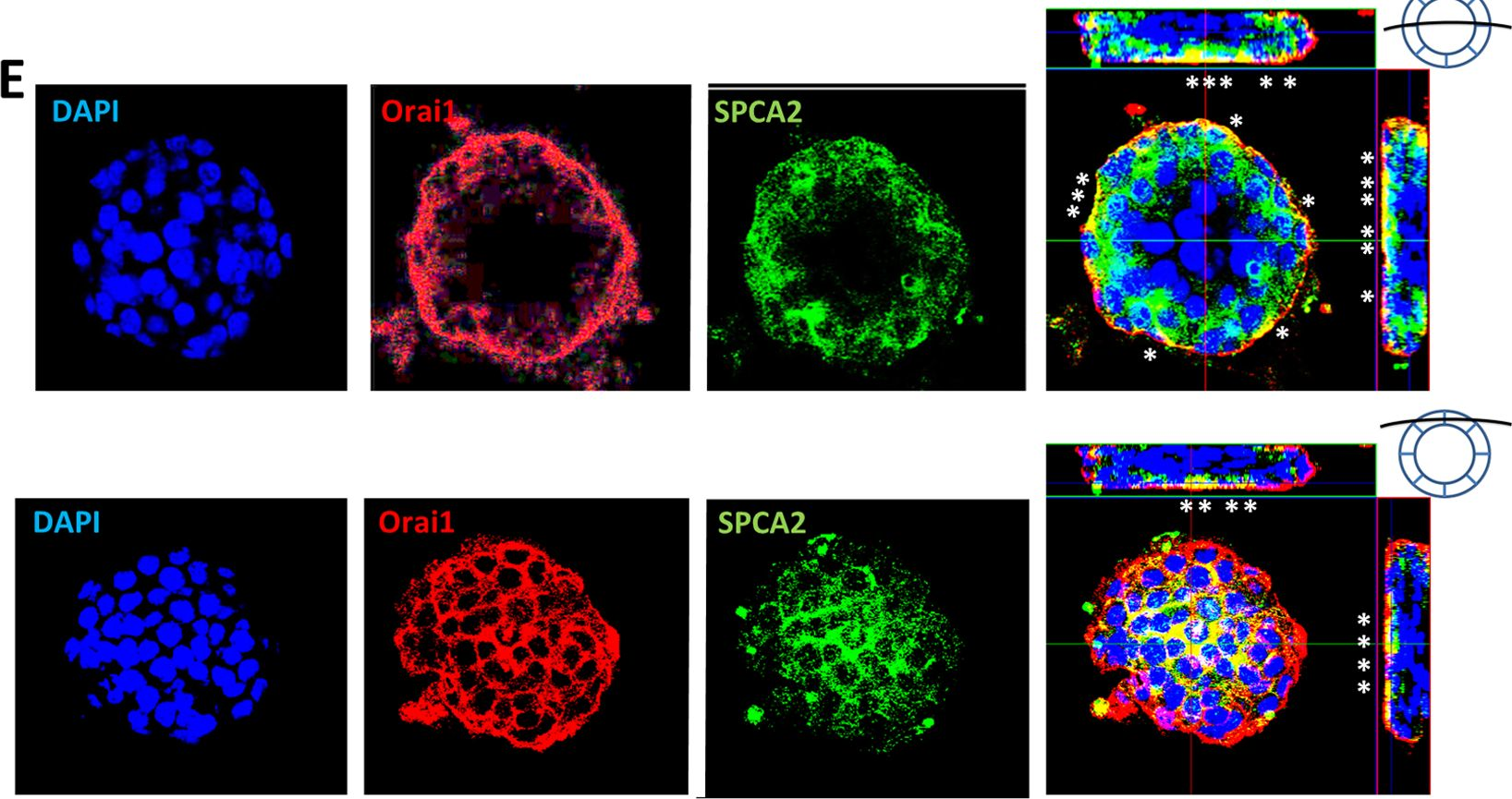
-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
Fig.5.C
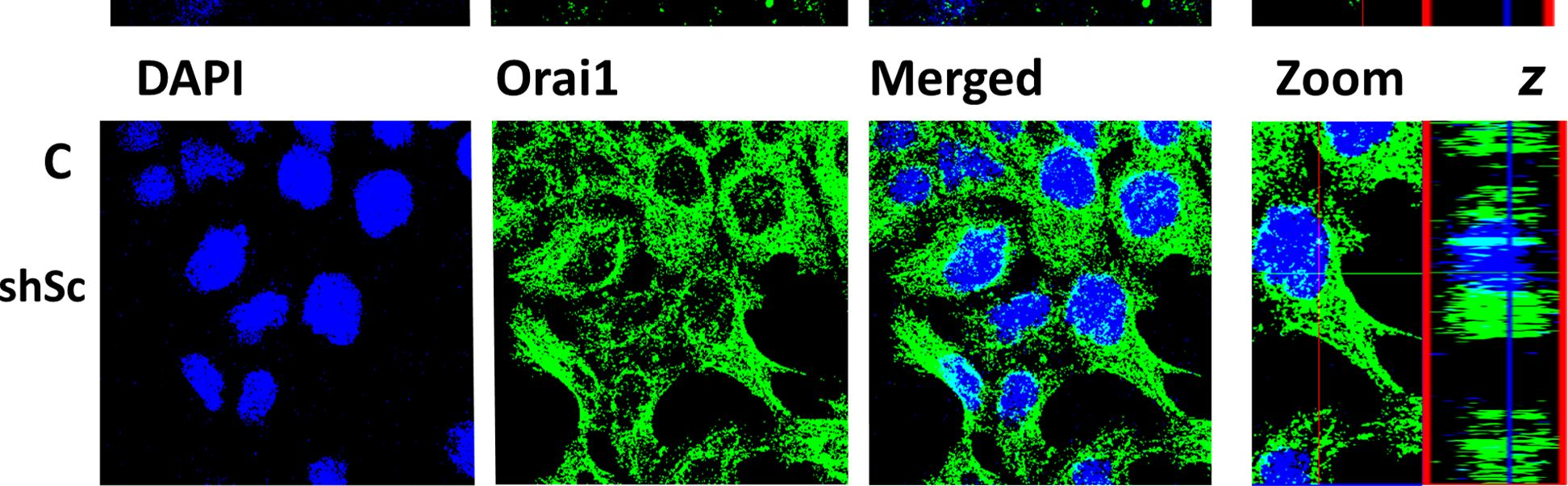
-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 11 July 2013 by Cross, B. M., Hack, A., et al.
Fig.5.D
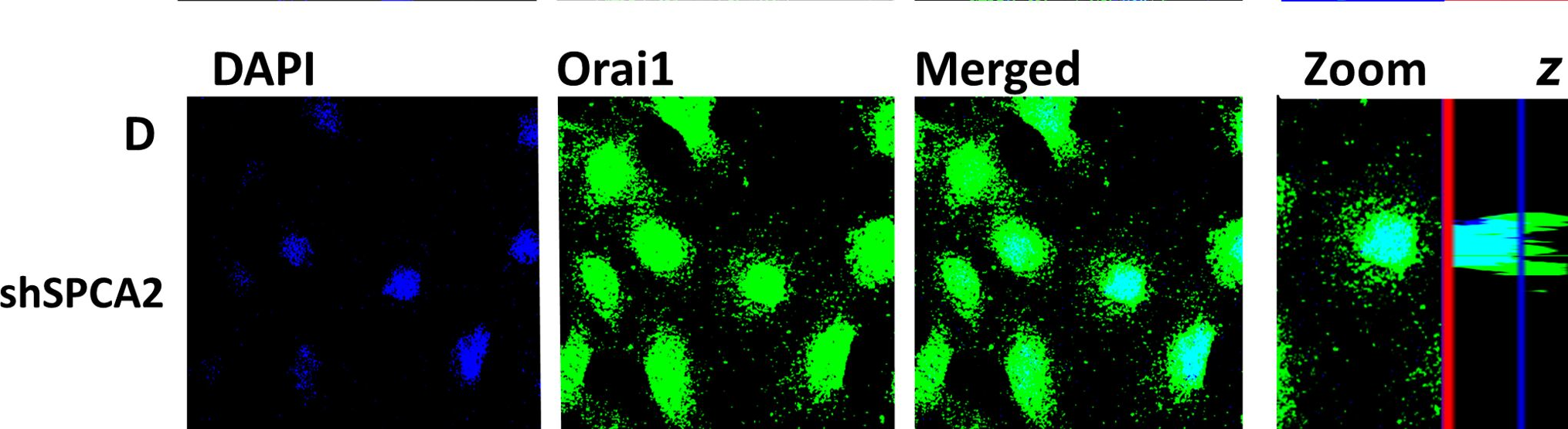
-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 7