White adipose tissue (WAT) fibrosis occurring in obesity contributes to the inflammatory and metabolic co-morbidities of insulin resistance and type 2 diabetes, yet the mechanisms involved remain poorly understood. Here, we report a role for the broadly conserved microRNA miR-30a as a regulator of WAT fibrosis and systemic glucose metabolism. Mice modified to express miR-30a at elevated levels in adipose tissues maintain insulin sensitivity coupled with reduced fatty liver disease when fed high fat diet. These effects were attributable to cell-autonomous functions of miR-30a that potently increase expression of adipocyte-specific genes. Proteomic screening revealed miR-30a limits pro-fibrotic programs in subcutaneous WAT, at least in part, by repressing PAI-1, a dominant regulator of fibrinolysis and biomarker of insulin resistance. Conversely, mouse adipocytes lacking miR-30a exhibited greater expression of fibrosis markers with disrupted cellular metabolism. Lastly, miR-30a expression negatively correlates with PAI-1 levels in subcutaneous WAT from people with obesity, further supporting an anti-fibrotic role for miR-30a. Together, these findings uncover miR-30a as a critical regulator of adipose tissue fibrosis that predicts metabolically healthy obesity in people and mice.
Product Citations: 269
The microRNA miR-30a blocks adipose tissue fibrosis accumulation in obesity.
In The Journal of Clinical Investigation on 5 June 2025 by Saha, P. K., Sharp, R., et al.
Preprint on BioRxiv : the Preprint Server for Biology on 19 October 2024 by Kondo, Y., Koga, J., et al.
Background Ischemic heart disease is a leading cause of death worldwide, and heart failure after myocardial infarction (MI) is a growing issue in this aging society. Macrophages play central roles in left ventricular (LV) remodeling after MI. Mitochondria consistently change their morphology, including fission and fusion, but the role of these mitochondrial morphological changes, especially in macrophages, is unknown. This study aims to illuminate the effects and mechanisms of Dynamin-related protein 1 (Drp1), a molecule mediating mitochondrial fission, in macrophages for cardiac remodeling after MI. Methods This study utilized genetically altered mice lacking Drp1 in monocytes/macrophages (Drp1-KO) to elucidate the specific role of macrophage Drp1 in post-infarct LV remodeling. Results Deletion of Drp1 in macrophages exacerbated LV remodeling, including reduced ejection fraction and increased LV diameters, which resulted in decreased survival after MI. Histological analysis indicated increased fibrosis and sustained macrophage accumulation in Drp1-KO mice. Blockade of Drp1 in macrophages decreased mitochondrial fission and impaired mitophagy, leading to the subsequent release of mitochondrial DNA (mtDNA) to the cytosol and induction of inflammatory cytokines. This induction was abrogated by an autophagy inducer, Tat-beclin1, or siRNA-mediated knockdown of Z-DNA Binding Protein 1 (ZBP1). Deletion of ZBP1 in bone marrow-derived cells abrogated LV remodeling induced by Drp1 inhibitor, Mdivi-1. Conclusion Macrophage Drp1 plays a critical role in the pathobiology of LV remodeling after MI, especially mitochondria quality control mechanisms. Macrophage Drp1 could be a novel therapeutic molecule to mitigate the progression of LV remodeling and consequent heart failure after MI.
-
Mus musculus (House mouse)
-
Cardiovascular biology
-
Cell Biology
-
Immunology and Microbiology
A New Ex Vivo Model to Study Cardiac Fibrosis in Whole Mouse Hearts.
In JACC. Basic To Translational Science on 1 August 2024 by Kruithof, B. P. T., Mousavi Gourabi, B., et al.
Fibrosis is a characteristic of many cardiac diseases for which no effective treatment exists. We have developed an ex vivo flow system, which allows induction of cardiac fibrosis in intact adult mouse hearts. Lineage-tracing studies indicated that the collagen-producing myofibroblasts originated from the resident fibroblasts. The extent of fibrosis was flow rate dependent, and pharmacological inhibition of the transforming growth factor beta signaling pathway prevented fibrosis. Therefore, in this powerful system, the cellular and molecular mechanisms underlying cardiac fibrosis can be studied. In addition, new targets can be tested on organ level for their ability to inhibit fibrosis.
© 2024 The Authors.
-
Cardiovascular biology
DHCR24 inhibitor SH42 increases desmosterol without preventing atherosclerosis development in mice.
In IScience on 21 June 2024 by Ge, X., Slütter, B., et al.
The liver X receptor (LXR) is considered a therapeutic target for atherosclerosis treatment, but synthetic LXR agonists generally also cause hepatic steatosis and hypertriglyceridemia. Desmosterol, a final intermediate in cholesterol biosynthesis, has been identified as a selective LXR ligand that suppresses inflammation without inducing lipogenesis. Δ24-Dehydrocholesterol reductase (DHCR24) converts desmosterol into cholesterol, and we previously showed that the DHCR24 inhibitor SH42 increases desmosterol to activate LXR and attenuate experimental peritonitis and metabolic dysfunction-associated steatotic liver disease. Here, we aimed to evaluate the effect of SH42 on atherosclerosis development in APOE∗3-Leiden.CETP mice and low-density lipoproteins (LDL) receptor knockout mice, models for lipid- and inflammation-driven atherosclerosis, respectively. In both models, SH42 increased desmosterol without affecting plasma lipids. While reducing liver lipids in APOE∗3-Leiden.CETP mice, and regulating populations of circulating monocytes in LDL receptor knockout mice, SH42 did not attenuate atherosclerosis in either model.
© 2024 The Author(s).
-
Mus musculus (House mouse)
Attenuation of A(H7N9) influenza virus infection in mice exposed to cigarette smoke.
In Npj Viruses on 25 March 2024 by Fukuyama, S., Shoemaker, J. E., et al.
Influenza A(H7N9) virus showed high pathogenicity in humans when it emerged in 2013. Cigarette smoke (CS) causes pulmonary diseases including bronchitis, emphysema, and lung cancer. Although habitual smoking is thought to increase the risk of severe seasonal influenza virus infection, its effect on A(H7N9) virus infection is poorly understood. Here, we employed a mouse model of long-term exposure to CS to investigate the effect of CS on the pathogenicity of A(H7N9) virus infection. Unexpectedly, body weight loss for mice exposed to CS was milder than that for mock-treated mice upon A(H7N9) virus infection. CS exposure improved the survival rate of A(H7N9) virus-infected mice even though virus titers and pathological changes in the lungs were not significantly different between CS-exposed and control mice. Microarray analysis showed that CS-exposure activates cytokine/chemokine activity, immune response, and cell cycle activities that resemble reactivities against A(H7N9) virus infection. Therefore, under conditions where cytokine and chemokine expression in the lungs is already high due to CS exposure, the enhanced expression of cytokines and chemokines caused by A(H7N9) virus infection might be less harmful to the organs compared to the rapid increase in cytokine and chemokine expression in the air-exposed mice due to the infection. CS may thus induce immunoregulatory effects that attenuate severe pulmonary disease during A(H7N9) virus infection. However, these findings do not support CS exposure due to its many other proven negative health effects.
© 2024. The Author(s).
-
Immunology and Microbiology
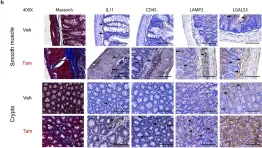
In PLoS One on 10 January 2020 by Lim, W. W., Ng, B., et al.
Fig.2.H

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 4
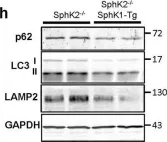
In Sci Rep on 4 December 2019 by Ishimaru, K., Yoshioka, K., et al.
Fig.5.H

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 4
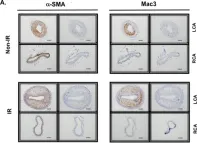
In Front Cardiovasc Med on 21 April 2018 by Ko, K. A., Wang, Y., et al.
Fig.6.A

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from Front Cardiovasc Med by CiteAb, provided under a CC-BY license
Image 1 of 4
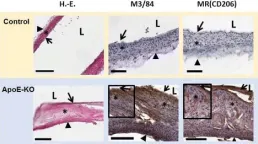
In EJNMMI Res on 1 December 2017 by Varasteh, Z., Hyafil, F., et al.
Fig.5.A

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from EJNMMI Res by CiteAb, provided under a CC-BY license
Image 1 of 4