Atherosclerotic lesions develop preferentially in arterial regions exposed to disturbed blood flow, where endothelial cells acquire an inflammatory phenotype. How disturbed flow induces endothelial cell inflammation is incompletely understood. Here we show that histone H3.3 phosphorylation at serine 31 (H3.3S31) regulates disturbed-flow-induced endothelial inflammation by allowing rapid induction of FOS and FOSB, required for inflammatory gene expression. We identified protein kinase N1 (PKN1) as the kinase responsible for disturbed-flow-induced H3.3S31 phosphorylation. Disturbed flow activates PKN1 in an integrin α5β1-dependent manner and induces its translocation into the nucleus, and PKN1 is also involved in the phosphorylation of the AP-1 transcription factor JUN. Mice with endothelium-specific PKN1 loss or endothelial expression of S31 phosphorylation-deficient H.3.3 mutants show reduced endothelial inflammation and disturbed-flow-induced vascular remodeling in vitro and in vivo. Together, we identified a pathway whereby disturbed flow through PKN1-mediated histone phosphorylation and FOS/FOSB induction promotes inflammatory gene expression and vascular inflammation.
© 2025. The Author(s).
Product Citations: 61
In Nat Cardiovasc Res on 1 February 2025 by Jin, Y. J., Liang, G., et al.
-
IHC
-
Genetics
Ependymal cell lineage reprogramming as a potential therapeutic intervention for hydrocephalus.
In EMBO Molecular Medicine on 1 November 2024 by Kaplani, K., Lalioti, M. E., et al.
Hydrocephalus is a common neurological condition, characterized by the excessive accumulation of cerebrospinal fluid in the cerebral ventricles. Primary treatments for hydrocephalus mainly involve neurosurgical cerebrospinal fluid diversion, which hold high morbidity and failure rates, highlighting the necessity for the discovery of novel therapeutic approaches. Although the pathophysiology of hydrocephalus is highly multifactorial, impaired function of the brain ependymal cells plays a fundamental role in hydrocephalus. Here we show that GemC1 and McIdas, key regulators of multiciliated ependymal cell fate determination, induce direct cellular reprogramming towards ependyma. Our study reveals that ectopic expression of GemC1 and McIdas reprograms cortical astrocytes and programs mouse embryonic stem cells into ependyma. McIdas is sufficient to establish functional activity in the reprogrammed astrocytes. Furthermore, we show that McIdas' expression promotes ependymal cell regeneration in two different postnatal hydrocephalus mouse models: an intracranial hemorrhage and a genetic form of hydrocephalus and ameliorates the cytoarchitecture of the neurogenic niche. Our study provides evidence on the restoration of ependyma in animal models mimicking hydrocephalus that could be exploited towards future therapeutic interventions.
© 2024. The Author(s).
-
Biochemistry and Molecular biology
-
Neuroscience
-
Stem Cells and Developmental Biology
In Proceedings of the National Academy of Sciences of the United States of America on 30 July 2024 by Weaver, O., Gano, D., et al.
Cerebellar injury in preterm infants with central nervous system (CNS) hemorrhage results in lasting neurological deficits and an increased risk of autism. The impact of blood-induced pathways on cerebellar development remains largely unknown, so no specific treatments have been developed to counteract the harmful effects of blood after neurovascular damage in preterm infants. Here, we show that fibrinogen, a blood-clotting protein, plays a central role in impairing neonatal cerebellar development. Longitudinal MRI of preterm infants revealed that cerebellar bleeds were the most critical factor associated with poor cerebellar growth. Using inflammatory and hemorrhagic mouse models of neonatal cerebellar injury, we found that fibrinogen increased innate immune activation and impeded neurogenesis in the developing cerebellum. Fibrinogen inhibited sonic hedgehog (SHH) signaling, the main mitogenic pathway in cerebellar granule neuron progenitors (CGNPs), and was sufficient to disrupt cerebellar growth. Genetic fibrinogen depletion attenuated neuroinflammation, promoted CGNP proliferation, and preserved normal cerebellar development after neurovascular damage. Our findings suggest that fibrinogen alters the balance of SHH signaling in the neurovascular niche and may serve as a therapeutic target to mitigate developmental brain injury after CNS hemorrhage.
-
WB
-
Mus musculus (House mouse)
-
Cardiovascular biology
In The Journal of Clinical Investigation on 21 May 2024 by Hong, S. G., Ashby, J. W., et al.
Endothelial cells (ECs) in the descending aorta are exposed to high laminar shear stress, and this supports an anti-inflammatory phenotype. High laminar shear stress also induces flow-aligned cell elongation and front-rear polarity, but whether these are required for the anti-inflammatory phenotype is unclear. Here, we showed that Caveolin-1-rich microdomains polarize to the downstream end of ECs that are exposed to continuous high laminar flow. These microdomains were characterized by high membrane rigidity, filamentous actin (F-actin), and raft-associated lipids. Transient receptor potential vanilloid-type 4 (TRPV4) ion channels were ubiquitously expressed on the plasma membrane but mediated localized Ca2+ entry only at these microdomains where they physically interacted with clustered Caveolin-1. These focal Ca2+ bursts activated endothelial nitric oxide synthase (eNOS) within the confines of these domains. Importantly, we found that signaling at these domains required both cell body elongation and sustained flow. Finally, TRPV4 signaling at these domains was necessary and sufficient to suppress inflammatory gene expression, and exogenous activation of TRPV4 channels ameliorated the inflammatory response to stimuli both in vitro and in vivo. Our work revealed a polarized mechanosensitive signaling hub in arterial ECs that dampens inflammatory gene expression and promotes cell resilience.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In Science Advances on 16 February 2024 by Cavallero, S., Roustaei, M., et al.
Exercise promotes pulsatile shear stress in the arterial circulation and ameliorates cardiometabolic diseases. However, exercise-mediated metabolic transducers for vascular protection remain under-investigated. Untargeted metabolomic analysis demonstrated that wild-type mice undergoing voluntary wheel running exercise expressed increased endothelial stearoyl-CoA desaturase 1 (SCD1) that catalyzes anti-inflammatory lipid metabolites, namely, oleic (OA) and palmitoleic acids (PA), to mitigate NF-κB-mediated inflammatory responses. In silico analysis revealed that exercise augmented time-averaged wall shear stress but mitigated flow recirculation and oscillatory shear index in the lesser curvature of the mouse aortic arch. Following exercise, endothelial Scd1-deleted mice (Ldlr-/- Scd1EC-/-) on high-fat diet developed persistent VCAM1-positive endothelium in the lesser curvature and the descending aorta, whereas SCD1 overexpression via adenovirus transfection mitigated endoplasmic reticulum stress and inflammatory biomarkers. Single-cell transcriptomics of the aorta identified Scd1-positive and Vcam1-negative endothelial subclusters interacting with other candidate genes. Thus, exercise mitigates flow recirculation and activates endothelial SCD1 to catalyze OA and PA for vascular endothelial protection.
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Cell Biology
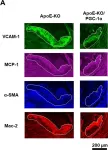
In Sci Rep on 11 March 2019 by Shimba, Y., Togawa, H., et al.
Fig.4.A

-
IHC-Frozen
-
Mus musculus (House mouse)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 4
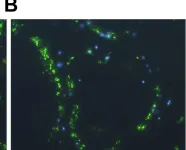
In PLoS Pathog on 19 September 2008 by Stanley, A. C., Dalton, J. E., et al.
Fig.6.B

-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 4
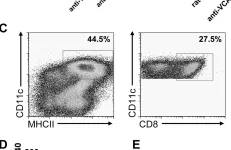
In PLoS Pathog on 19 September 2008 by Stanley, A. C., Dalton, J. E., et al.
Fig.7.C

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 4
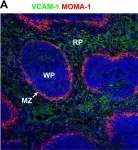
In PLoS Pathog on 19 September 2008 by Stanley, A. C., Dalton, J. E., et al.
Fig.5.A

-
IHC-P-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 4