

Cardiotoxicity caused by immune checkpoint inhibitors is one of the most severe and potentially fatal side effects. Hence it is crucial from a therapeutic standpoint to understand the underlying processes and devise countermeasures. This study sought to determine whether the SOCS3/JAK/STAT3 signaling pathway, which controls macrophage polarization, contributes to the cardiotoxicity caused by PD-1/PD-L1 inhibitors. The PD-1/PD-L1 inhibitor BMS-1 (10 mg/kg) was used to create a mouse model of immune checkpoint inhibitor-related cardiotoxicity, and hematoxylin and Masson's trichome tests were used to measure cardiomyocyte apoptosis and cardiotoxicity. The production of M1 factors (tumor necrosis factor α [TNF-α] and interleukin [IL]-1 b), as well as the blood levels of myocardial enzymes (creatine kinase, aspartate transaminase, creatine kinase-MB, and lactate dehydrogenase), were evaluated by ELISA. Echocardiography was used to assess the heart's health. The processes were investigated using flow cytometric analysis, real-time PCR, Western blot, and chromatin immunoprecipitation. We found that the PD-1/PD-L1 inhibitor BMS-1 dramatically reduced tumor weight while considerably impairing cardiac function in melanoma-induced tumor-bearing mice. At the gene and protein levels, it was found that levels of SOCS3, JAK, STAT3, and the inflammatory mediators IL-6 and TNF-α had all significantly decreased. Immune checkpoint inhibitor-induced cardiotoxicity may be linked to major changes in the SOCS3/JAK/STAT3 signaling pathway, as indicated by the knockdown of SOCS3, JAK, and STAT3. Finally, immune checkpoint inhibitor intervention demonstrated a large elevation of CD86+ and MHCII+ as well as a considerable increase in macrophages. These data suggest that the SOCS3/JAK/STAT3 signaling pathway, which controls macrophage polarization, may be linked to cardiotoxicity caused by PD-1/PD-L1 inhibitor therapy.
Copyright © 2025 Termedia.
Product Citations: 217
In Central-European Journal of Immunology / Polish Society for Immunology and Eleven Other Central-European Immunological Societies on 7 July 2025 by Fu, J., Wang, G., et al.
-
Immunology and Microbiology
In Cell Death & Disease on 6 February 2025 by Peng, W., Qin, Q., et al.
Sepsis is a life-threatening condition characterized by a dysregulated immune response to infection, leading to systemic inflammation and organ dysfunction. Macrophage polarization plays a critical role in pathogenesis of sepsis, and the influence of B lymphocyte-induced maturation protein-1 (Blimp-1) on this polarization is an underexplored yet pivotal aspect. This study aimed to elucidate the role of Blimp-1 in macrophage polarization and metabolism during sepsis. Using a murine cecal ligation and puncture model, we observed elevated Blimp-1 expression in M2 macrophages. Knockdown of Blimp-1 by macrophage-targeted adeno-associated virus in this model resulted in decreased survival rates, exacerbated tissue damage, and impaired M2 polarization, underscoring its protective role in sepsis. In vitro studies with bone marrow-derived macrophage (BMDM), RAW264.7, and THP-1 cells further demonstrated Blimp-1 promotes M2 polarization and modulates key metabolic pathways. Metabolomics and dual-luciferase assays revealed Blimp-1 significantly influences purine biosynthesis and the downstream Ornithine cycle, which are essential for M2 macrophage polarization. In vitro studies with BMDM further suggested that the purine biosynthesis and Ornithine cycle metabolic regulation is involved in Blimp-1's effects on M2 macrophage polarization, and mediates Blimp-1's impact on septic mice. Our findings unveil a novel mechanism by which Blimp-1 modulates macrophage polarization through metabolic regulation, presenting potential therapeutic targets for sepsis. This study highlights the significance of Blimp-1 in orchestrating macrophage responses and metabolic adaptations in sepsis, offering valuable insights into its role as a critical regulator of immune and metabolic homeostasis.
© 2025. The Author(s).
-
Biochemistry and Molecular biology
-
Cell Biology
-
Genetics
-
Immunology and Microbiology
Epidermal maintenance of Langerhans cells relies on autophagy-regulated lipid metabolism.
In The Journal of Cell Biology on 3 February 2025 by Arbogast, F., Sal-Carro, R., et al.
Macroautophagy (often-named autophagy), a catabolic process involving autophagy-related (Atg) genes, prevents the accumulation of harmful cytoplasmic components and mobilizes energy reserves in long-lived and self-renewing cells. Autophagy deficiency affects antigen presentation in conventional dendritic cells (DCs) without impacting their survival. However, previous studies did not address epidermal Langerhans cells (LCs). Here, we demonstrate that deletion of either Atg5 or Atg7 in LCs leads to their gradual depletion. ATG5-deficient LCs showed metabolic dysregulation and accumulated neutral lipids. Despite increased mitochondrial respiratory capacity, they were unable to process lipids, eventually leading them to ferroptosis. Finally, metabolically impaired LCs upregulated proinflammatory transcripts and showed decreased expression of neuronal interaction receptors. Altogether, autophagy represents a critical regulator of lipid storage and metabolism in LCs, allowing their maintenance in the epidermis.
© 2024 Arbogast et al.
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Cell Biology
Preprint on BioRxiv : the Preprint Server for Biology on 22 January 2025 by Eislmayr, K. D., Langner, C., et al.
Bacteria of the genus Shigella replicate in intestinal epithelial cells and cause shigellosis, a severe diarrheal disease that resolves spontaneously in most healthy individuals. During shigellosis, neutrophils are abundantly recruited to the gut, and have long been thought to be central to Shigella control and pathogenesis. However, how shigellosis resolves remains poorly understood due to the longstanding lack of a tractable and physiological animal model. Here, using our newly developed Nlrc4 −/− Casp11 −/− mouse model of shigellosis, we unexpectedly find no major role for neutrophils in limiting Shigella or in disease pathogenesis. Instead, we uncover an essential role for macrophages in the host control of Shigella . Macrophages respond to Shigella via TLRs to produce IL-12, which then induces IFN-γ, a cytokine that is essential to control Shigella replication in intestinal epithelial cells. Collectively, our findings reshape our understanding of the innate immune response to Shigella .
-
Mus musculus (House mouse)
In EBioMedicine on 13 December 2024 by Hu, H., Li, X., et al.
Lung metastasis is a critical and often fatal progression in cancer patients, with monocyte-derived macrophages (Mo-macs) playing multifaceted roles in this process. Despite the recognized importance of Mac-macs, most studies focus on these cells themselves, while the precise mechanisms through which tumor cells manipulate Mo-macs to promote metastasis remain poorly understood.
We developed an in vivo CRISPR screening system to identify genes involved in macrophage-dependent metastasis by depleting Mo-macs. Osteoprotegerin (OPG) was identified as the factor significantly enhances lung metastasis. We validated its function in lung metastasis by modulating the expression of OPG in an array of cell lines and performed spontaneous and experimental lung metastasis assays. Genetically engineered mice were utilized to confirm the role of RANKL-RANK signaling in OPG-mediated metastasis. Additionally, we employed different neutralizing antibodies to elucidate the roles of Mo-macs and NK cells and inhibitor to clarify the role of CXCL10 signaling.
Employing in vivo screening techniques, we elucidate the role of OPG, a protein secreted by cancer cells, in driving lung metastasis, contingent upon regulating Mo-mac activity. OPG blocks the signaling cascade between receptor activator of nuclear factor kappa-B ligand (RANKL) and its receptor RANK on Mo-macs, thereby hindering Mo-macs from secreting CXCL10, a chemokine crucial for recruiting natural killer (NK) cells that help control lung metastasis. Moreover, we observe an enrichment of OPG amplifications in metastatic cancer patients, and elevated levels of OPG expression in lung metastatic sites compared to paired primary breast cancer samples.
Our work revealed that OPG works as a lung metastasis promoting factor by blocking the RANKL-RANK-CXCL10 axis to drive the paucity of NK cells, which could be a therapeutic target for lung metastatic cancer patients.
The full list of funding supporting this study can be found in the Acknowledgements section.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
-
Cancer Research
In Oxid Med Cell Longev on 6 July 2017 by Ramadori, P., Drescher, H., et al.
Fig.6.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Oxid Med Cell Longev by CiteAb, provided under a CC-BY license
Image 1 of 7
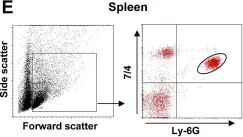
In PLoS Pathog on 1 March 2014 by Di Paolo, N. C., Baldwin, L. K., et al.
Fig.1.E

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 7
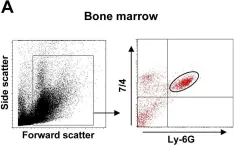
In PLoS Pathog on 1 March 2014 by Di Paolo, N. C., Baldwin, L. K., et al.
Fig.1.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 7
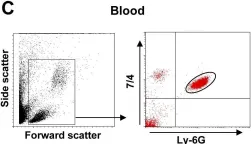
In PLoS Pathog on 1 March 2014 by Di Paolo, N. C., Baldwin, L. K., et al.
Fig.1.C

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 7
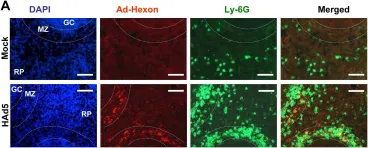
In PLoS Pathog on 1 March 2014 by Di Paolo, N. C., Baldwin, L. K., et al.
Fig.3.A

-
IHC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS Pathog on 1 March 2014 by Di Paolo, N. C., Baldwin, L. K., et al.
Fig.3.C

-
IHC-IF
-
Mus musculus (House mouse)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 7
In J Neuroinflammation on 16 January 2012 by Lee, E., Chanamara, S., et al.
Fig.4.A

-
IHC-IF
-
Mus musculus (House mouse)
Collected and cropped from J Neuroinflammation by CiteAb, provided under a CC-BY license
Image 1 of 7