





The transmission pattern of mpox has shifted from sporadic zoonotic outbreaks to sustained human-to-human spread. Epidemiological data indicate sexual contact as a crucial driver for efficient transmission and the associated devastating mpox outbreaks in recent years. However, our understanding of exact driving factors and transmission determinants is still limited. Here, we investigate MPXV clade Ia virus pathogenesis, shedding kinetics, and transmission potential in a prairie dog model (Cynomys ludovicianus). Mucosal inoculation via all routes (penile/preputial, vaginal, rectal, intranasal) results in a productive, systemic infection. Inoculation via urogenital routes generate the highest virus shedding and most severe clinical disease. Simulated sexual contact transmission results in 100% transmission efficiency with high virus shedding in sentinels on day 1, even before the onset of visible clinical signs. Our findings provide experimental support for the importance of anogenital mucosal infection and transmission and establish a model for studying these in vivo. These results advocate for a stronger focus on mucosal infection when evaluating countermeasures.
© 2026. The Author(s).
Product Citations: 293
In Nature Communications on 25 June 2026 by Kaiser, F. K., Mukesh, R. K., et al.
-
FC/FACS
-
Canis lupus familiaris (Domestic dog)
-
Immunology and Microbiology
In Journal of Translational Medicine on 22 June 2026 by Liu, Y., Guo, Q., et al.
Radiotherapy (RT) resistance remains a significant challenge in esophageal squamous cell carcinoma (ESCC). While Nrf2 is known to mediate antioxidant defense, its role in modulating the immunogenicity of radiotherapy-induced cell death is poorly understood.
We established a spontaneous esophageal cancer model using genetically engineered mice with conditional knockout of Nrf2, and analyzed the tumor immune microenvironment by single-cell RNA sequencing. In vitro, we performed co-culture experiments using CRISPR/Cas9-mediated Nrf2-knockout esophageal cancer cell lines with dendritic cells and T cells to validate immune activation. In addition, in vivo validation was conducted using a mouse subcutaneous tumor model.
ScRNA-seq revealed that Nrf2 deficiency significantly remodeled the myeloid compartment, characterized by a population shift from Folr2 + to Mrc1 + macrophages and an expansion of effector T cells. Mechanistically, Nrf2 deletion downregulated the expression of Gpx2, impairing antioxidant defenses. This sensitized ESCC cells to RT, triggering the release of immunogenic cell death (ICD) markers, including ATP, HMGB1, and surface calreticulin (CRT). In tumor-DC-T cell co-culture systems, Nrf2-deficient cells stimulated dendritic cells (DCs) to secrete IP-10 (CXCL10), which was indispensable for the recruitment and activation of CD8 + T cells. Finally, in vivo experiments confirmed that Nrf2 deficiency enhanced radiosensitivity and promoted CD8 + T cell infiltration.
Our findings identify the Nrf2-Gpx2 axis as a master regulator of immunogenicity in ESCC. Targeting this axis represents a promising strategy to convert "cold" tumors into "hot" environments, thereby improving the efficacy of radiotherapy and immunotherapy.
© 2026. The Author(s).
-
FC/FACS
-
Mus musculus (House mouse)
Preprint on Research Square on 6 June 2026 by Todd, B. P., Luo, Z., et al.
Abstract Type I interferon (IFN-I) signaling has emerged as a central regulator of neuroinflammation across diverse central nervous system disorders, including traumatic brain injury (TBI). While TBI is a leading cause of neurologic morbidity and mortality through young adulthood, there is a paucity of neuroprotective therapies available to clinicians. Recent work has demonstrated neuroprotection after global IFN-I deficiency, yet the cell-type-specific contributions to traumatic brain injury (TBI) and the mechanisms of immune modulation remain poorly defined. Using mice with microglia-specific IFN-I receptor deficiency, we show that loss of microglial IFN-I responsiveness suppresses microglial reactivity, reducing microglial accumulation, synaptic engulfment, antigen presentation, and T cell interactions after TBI. This attenuation preserves neuronal integrity and limits thalamic neuronal loss. Despite this neuroprotection, microglia-restricted IFN-I blockade reveals functional redundancy across CNS cell types, underscoring the multi-cellular nature of IFN-I signaling in the injured brain. Together, our findings delineate a microglial IFN-I–dependent pathway that exacerbates secondary injury after TBI and highlight both the therapeutic potential and inherent limitations of cell-type-targeted IFN-I modulation.
-
FC/FACS
-
Mus musculus (House mouse)
-
Neuroscience
Mitophagy promotes lung repair and regeneration by restoring epithelial metabolic fitness.
In Nature Communications on 14 April 2026 by Wu, P., Chen, J., et al.
Alveolar Type II cells (AT2s) are the stem cells responsible for both lung homeostasis and regeneration. Mitochondrial dysfunction in AT2 cells has been implicated in both chronic and acute injury-induced alveolar diseases, including idiopathic pulmonary fibrosis (IPF) and viral pneumonia. However, the role of mitochondrial homeostasis in post-injury lung repair and regeneration remains elusive. Here we demonstrate that genetic depletion of Ubiquitin Specific Peptidase 30 (USP30), a negative regulator of mitophagy, boosts mitophagy and restores mitochondrial function in AT2 cells, leading to protection from injury-induced apoptosis and enhanced stem cell activity. Both global and AT2-specific Usp30 knockout (KO) promote alveolar regeneration, protecting the mice from bleomycin-induced lung fibrosis and influenza pneumonia. Moreover, pharmacological inhibition of USP30 effectively alleviates these conditions. Together, our findings reveal a previously underappreciated role for mitophagy in lung injury and repair and highlight USP30 inhibition as a promising therapeutic strategy for treating alveolar diseases.
© 2026. The Author(s).
-
FC/FACS
-
Mus musculus (House mouse)
-
Cell Biology
-
Biochemistry and Molecular biology
Adipocytes signal to recruit specific mRNAs from surrounding cells to restore expression deficits.
In Nature Communications on 11 April 2026 by Crewe, C., Gliniak, C. M., et al.
Extracellular vesicles (EVs) are nano-sized, membrane-delimited, particles released by cells that carry signaling macromolecules. A major pathway of EV production is potentiated by neutral sphingomyelinase 2 (SMPD3/nSMAse2), an enzyme that generates ceramide from sphingomyelin. In our attempt to study this pathway in adipocytes of male mice, we discover that the elimination of SMPD3 from adipocytes in vivo triggers a signal to surrounding immune cell-like preadipocytes to release EVs that carry SMPD3 mRNA. This results in a widespread increase in SMPD3 mRNA in purified null adipocytes without a change in the transcripts of other enzymes involved in ceramide metabolism. These results point to a selective mechanism by which specific mRNA molecules are acquired from the microenvironment to a level that can restore expression of mRNA and protein in a cell that is depleted of the corresponding genetic information.
© 2026. The Author(s).
-
FC/FACS
-
Mus musculus (House mouse)
-
Genetics
In Nat Commun on 7 October 2025 by Paladini, M. S., Yang, B. A., et al.
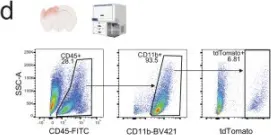
Fig.1.D
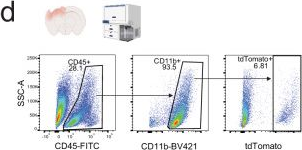
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 10
In Pharmaceutics on 28 March 2023 by Zhang, H., Li, F., et al.
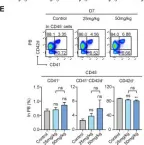
Fig.5.F
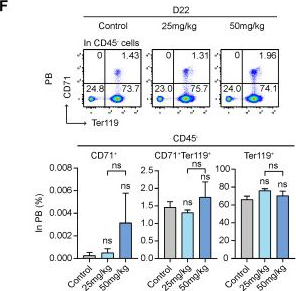
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Pharmaceutics by CiteAb, provided under a CC-BY license
Image 1 of 10
In Pharmaceutics on 28 March 2023 by Zhang, H., Li, F., et al.
Fig.5.C
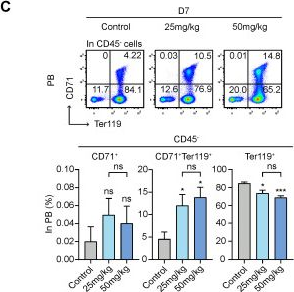
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Pharmaceutics by CiteAb, provided under a CC-BY license
Image 1 of 10
In Pharmaceutics on 28 March 2023 by Zhang, H., Li, F., et al.
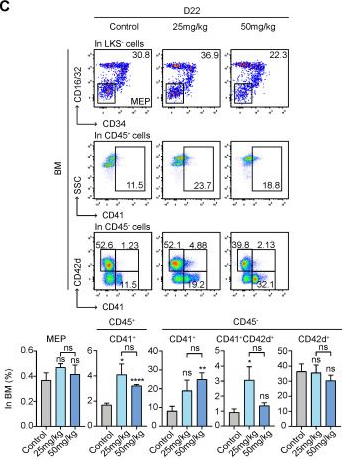
Fig.4.F
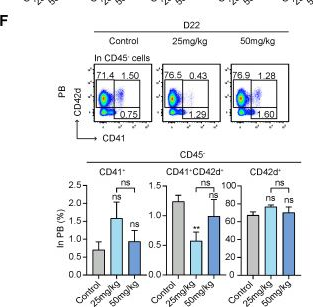
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Pharmaceutics by CiteAb, provided under a CC-BY license
Image 1 of 10
In Pharmaceutics on 28 March 2023 by Zhang, H., Li, F., et al.
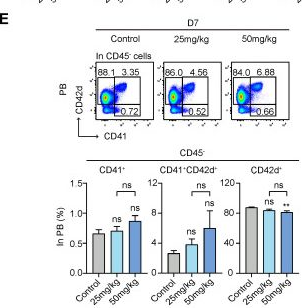
Fig.4.E
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Pharmaceutics by CiteAb, provided under a CC-BY license
Image 1 of 10
In Pharmaceutics on 28 March 2023 by Zhang, H., Li, F., et al.
Fig.4.C
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Pharmaceutics by CiteAb, provided under a CC-BY license
Image 1 of 10
In Nat Commun on 6 August 2022 by Jung, S. H., Hwang, B. H., et al.
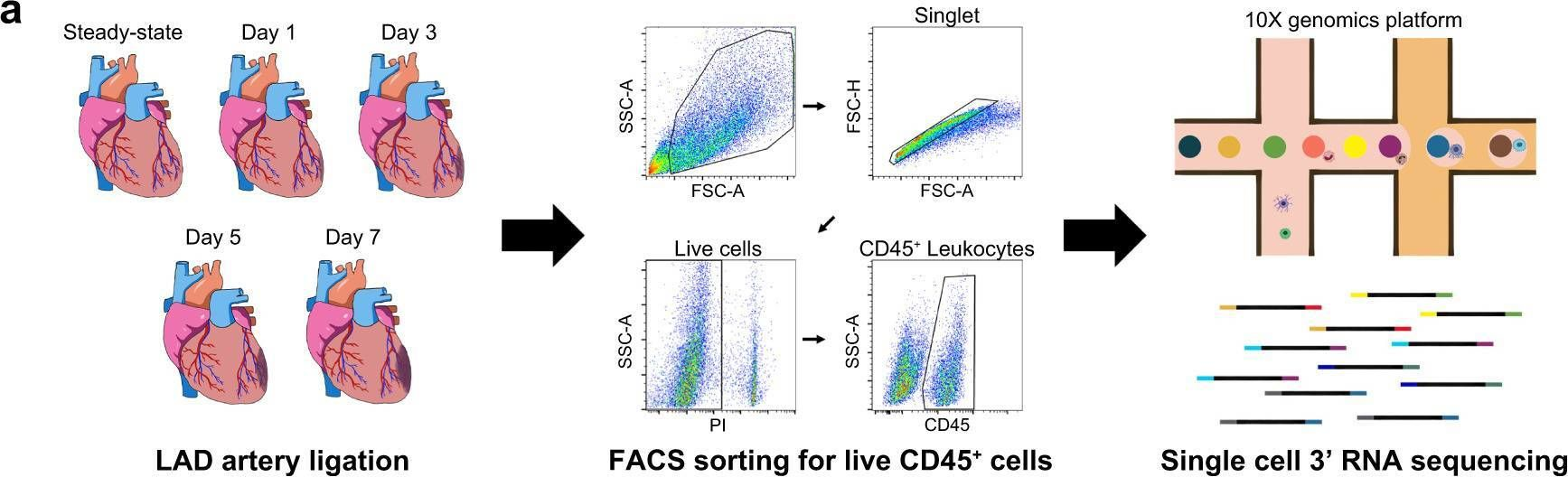
Fig.1.A
-
FC/FACS
-
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 10
In EMBO Mol Med on 8 June 2022 by Tisch, N., Mogler, C., et al.
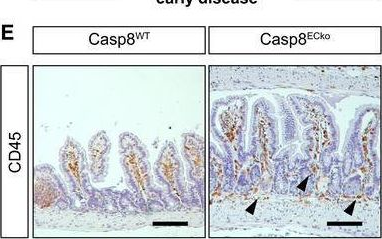
Fig.2.E
-
IHC
-
Mus musculus (House mouse)
Collected and cropped from EMBO Molecular Medicine by CiteAb, provided under a CC-BY license
Image 1 of 10
In Sci Rep on 5 June 2019 by Salas-Pérdomo, A., Miro-Mur, F., et al.
Fig.2.D
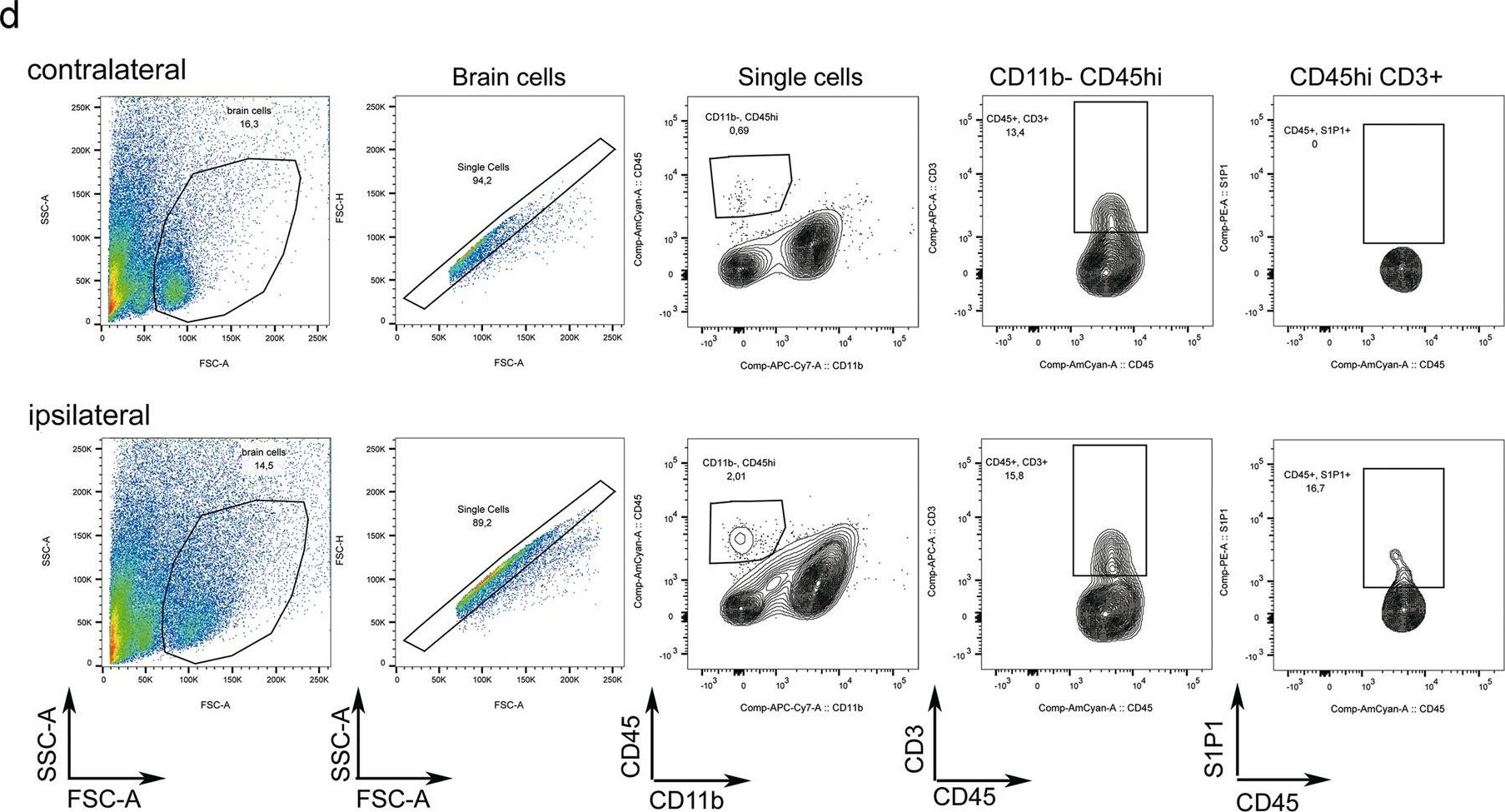
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Scientific Reports by CiteAb, provided under a CC-BY license
Image 1 of 10
In J Neuroinflammation on 2 September 2017 by Gerzanich, V., Makar, T. K., et al.
Fig.5.A
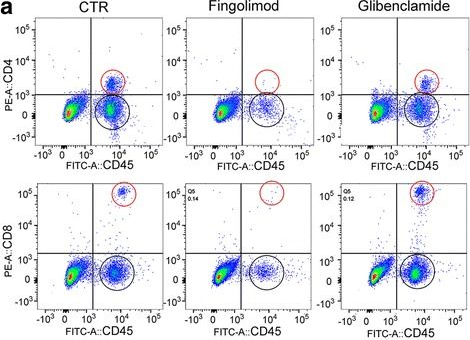
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Journal of Neuroinflammation by CiteAb, provided under a CC-BY license
Image 1 of 10