

Lentiviral vector (LV)-mediated ex vivo gene therapy for haematopoietic stem and progenitor cells (HSPCs) has delivered on the promise of a 'one-and-done' treatment for several genetic diseases1. However, ex vivo manipulation and patient conditioning before transplantation are major hurdles that could be overcome by an in vivo approach. Here we demonstrate that in vivo gene delivery to HSPCs after systemic LV administration is enabled by the substantial trafficking of these cells from the liver to the bone marrow in newborn mice. We improved gene-transfer efficiency using a phagocytosis-shielded LV, successfully reaching bona fide HSPCs capable of long-term multilineage output and engraftment after serial transplantation, as confirmed by clonal tracking. HSPC mobilization further increased gene transfer, extending the window of intervention, although permissiveness to LV transduction declined with age. We successfully tested this in vivo strategy in mouse models of adenosine deaminase deficiency, autosomal recessive osteopetrosis and Fanconi anaemia. Interestingly, in vivo gene transfer provided a selective advantage to corrected HSPCs in Fanconi anaemia, leading to near-complete haematopoietic reconstitution and prevention of bone marrow failure. Given that circulating HSPCs in humans are also most abundant shortly after birth, in vivo HSPC gene transfer holds strong translational potential across multiple diseases.
© 2025. The Author(s).
Product Citations: 81
In vivo haemopoietic stem cell gene therapy enabled by postnatal trafficking.
In Nature on 28 May 2025 by Milani, M., Fabiano, A., et al.
-
Stem Cells and Developmental Biology
In Acta Pharmaceutica Sinica. B on 1 March 2025 by Ouyang, Q., Cui, J., et al.
eIF3a is a N 6-methyladenosine (m6A) reader that regulates mRNA translation by recognizing m6A modifications of these mRNAs. It has been suggested that eIF3a may play an important role in regulating translation initiation via m6A during infection when canonical cap-dependent initiation is inhibited. However, the death of animal model studies impedes our understanding of the functional significance of eIF3a in immunity and regulation in vivo. In this study, we investigated the in vivo function of eIF3a using eIF3a knockout and knockdown mouse models and found that eIF3a deficiency resulted in splenic tissue structural disruption and multi-organ damage, which contributed to severe sepsis induced by Lipopolysaccharide (LPS). Ectopic eIF3a overexpression in the eIF3a knockdown mice rescued mice from LPS-induced severe sepsis. We further showed that eIF3a maintains a functional and healthy immune system by regulating B cell function and quantity through m6A modification of mRNAs. These findings unveil a novel mechanism underlying sepsis, implicating the pivotal role of B cells in this complex disease process regulated by eIF3a. Furthermore, eIF3a may be used to develop a potential strategy for treating sepsis.
© 2025 The Authors.
-
Immunology and Microbiology
In Nature Communications on 30 October 2024 by Li, W. P., Mao, X. T., et al.
T cell expansion has a crucial function in both autoimmune and chronic inflammatory diseases, with cycling T cells contributing to the pathogenesis of autoimmune diseases by causing uncontrolled immune responses and tissue damage. Yet the regulatory mechanisms governing T cell expansion remain incompletely understood. Here we show that the enzyme N-acetyltransferase 10 (NAT10) regulates T cell activation and proliferation upon antigen stimulation. T cell-specific NAT10 deficiency in mice reduces the number of mature T cells in peripheral lymphoid organs. Mechanistically, NAT10 acetylates RACK1 at K185, preventing subsequent RACK1 K48-linked ubiquitination and degradation. The increased RACK1 stability alters ribosome formation and cellular metabolism, leading to enhanced supply of energy and biosynthetic precursors and, eventually, T cell proliferation. Our findings thus highlight the essential function of NAT10 in T cell self-renewal and metabolism and elucidate NAT10 mode of action for the potential development of novel therapies for immune-related disorders.
© 2024. The Author(s).
-
Immunology and Microbiology
Preprint on BioRxiv : the Preprint Server for Biology on 17 October 2024 by Bi, H., Ren, K., et al.
Deleterious germline DDX41 variants constitute the most common inherited predisposition disorder linked to myeloid neoplasms (MNs). The role of DDX41 in hematopoiesis and how its germline and somatic mutations contribute to MNs remain unclear. Here we show that DDX41 is essential for erythropoiesis but dispensable for the development of other hematopoietic lineages. Using stage-specific Cre models for erythropoiesis, we reveal that Ddx41 knockout in early erythropoiesis is embryonically lethal, while knockout in late-stage terminal erythropoiesis allows mice to survive with normal blood counts. DDX41 deficiency induces a significant upregulation of G-quadruplexes (G4), noncanonical DNA structures that tend to accumulate in the early stages of erythroid precursors. We show that DDX41 co-localizes with G4 on the erythroid genome. DDX41 directly binds to and dissolves G4, which is significantly compromised in MN-associated DDX41 mutants. Accumulation of G4 by DDX41 deficiency induces erythroid genome instability, defects in ribosomal biogenesis, and upregulation of p53. However, p53 deficiency does not rescue the embryonic death of Ddx41 hematopoietic-specific knockout mice. In parallel, genome instability also activates the cGas-Sting pathway, which is detrimental to survival since cGas-deficient and hematopoietic-specific Ddx41 knockout mice are viable without detectable hematologic phenotypes, although these mice continue to show erythroid ribosomal defects and upregulation of p53. These findings are further supported by data from a DDX41 mutated MN patient and human iPSC-derived bone marrow organoids. Our study establishes DDX41 as a G4 dissolver, essential for erythroid genome stability and suppressing the cGAS-STING pathway.
In eLife on 5 September 2024 by Roumelioti, F. M., Tzaferis, C., et al.
miRNAs constitute fine-tuners of gene expression and are implicated in a variety of diseases spanning from inflammation to cancer. miRNA expression is deregulated in rheumatoid arthritis (RA); however, their specific role in key arthritogenic cells such as the synovial fibroblast (SF) remains elusive. Previous studies have shown that Mir221/222 expression is upregulated in RA SFs. Here, we demonstrate that TNF and IL-1β but not IFN-γ activated Mir221/222 gene expression in murine SFs. SF-specific overexpression of Mir221/222 in huTNFtg mice led to further expansion of SFs and disease exacerbation, while its total ablation led to reduced SF expansion and attenuated disease. Mir221/222 overexpression altered the SF transcriptional profile igniting pathways involved in cell cycle and ECM (extracellular matrix) regulation. Validation of targets of Mir221/222 revealed cell cycle inhibitors Cdkn1b and Cdkn1c, as well as the epigenetic regulator Smarca1. Single-cell ATAC-seq data analysis revealed increased Mir221/222 gene activity in pathogenic SF subclusters and transcriptional regulation by Rela, Relb, Junb, Bach1, and Nfe2l2. Our results establish an SF-specific pathogenic role of Mir221/222 in arthritis and suggest that its therapeutic targeting in specific subpopulations could lead to novel fibroblast-targeted therapies.
© 2024, Roumelioti et al.
-
FC/FACS
-
Mus musculus (House mouse)
In Elife on 5 September 2024 by Roumelioti, F. M., Tzaferis, C., et al.
Fig.3.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 3
In Elife on 5 September 2024 by Roumelioti, F. M., Tzaferis, C., et al.
Fig.3.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 3
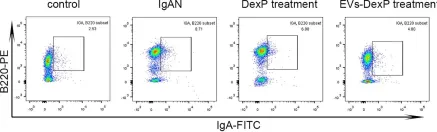
In Front Immunol on 20 September 2022 by Zhang, W., Yuan, Y., et al.
Fig.5.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Front Immunol by CiteAb, provided under a CC-BY license
Image 1 of 3