



Immune checkpoint blockades (ICBs) are promising, however they do not fit all types of tumor, such as those lack of tumor antigens. Induction of potent anti-tumor T cell immunity is critical for cancer therapy. In this study, we investigated the efficacy of immunotherapy via the immunogenic cell death (ICD) dying tumor cells in mouse models of lung metastasis and tumorigenesis.
ICD was induced by short exposure to lethal dose of chemotherapeutic drug doxorubicin (Dox), which initiated an irreversible ICD program in tumor cells. We immunized mice with ICD dying tumor cells in prevention, therapy in lung metastasis models, and Gprc5a-knockout (ko) model of lung tumorigenesis. T cells and macrophages isolated from lymph nodes or tumor tissues were analyzed by flow cytometry. Cytokines were analyzed by ELISA or Q-PCR analysis.
Immunization with these live but ICD dying tumor cells induced potent tumor-specific anti-tumor T cell immunity, which not only protected host from challenge by these tumor cells in prevention and therapy in mouse model of lung metastasis, but also prevented tumors development in Gprc5a-ko mouse model of lung tumorigenesis. The lymphocytes from lymph nodes and tumor tissues exhibited greatly enhanced activities of Th1 cells and M1 macrophages.
Immunization with the ICD dying tumor cells evokes potent tumor-specific T cell immunity, which provides a novel approach for cancer immunotherapy.
© 2025. The Author(s).
Product Citations: 172
In Journal of Cancer Research and Clinical Oncology on 18 January 2025 by Hu, M., Meng, X., et al.
-
Mus musculus (House mouse)
-
Cancer Research
-
Immunology and Microbiology
Preprint on BioRxiv : the Preprint Server for Biology on 2 January 2025 by Pastoors, D., Havermans, M., et al.
Enhancer translocations, due to 3q26 rearrangements, drive out-of-context MECOM expression in an aggressive subtype of acute myeloid leukemia (AML). Direct depletion of MECOM using an endogenous auxin-inducible degron immediately upregulates expression of myeloid differentiation factor CEBPA . MECOM depletion is also accompanied by a severe loss of stem cells and gain of differentiation. MECOM exerts its inhibitory effect by binding to the +42kb CEBPA enhancer, a gene essential for neutrophil development. This is partially dependent on the interaction between MECOM and its co-repressor CTBP2. We demonstrate that CEBPA overexpression can bypass the MECOM-mediated block of differentiation. In addition, AML patients with MECOM overexpression through enhancer hijacking show significantly reduced CEBPA . Our study directly connects two main players of myeloid transformation MECOM and CEBPA , and it provides insight into the mechanism by which MECOM maintains the stem cell state in a unique subtype of AML by inactivating CEBPA .
-
Cancer Research
PTPN1/2 inhibition promotes muscle stem cell differentiation in Duchenne muscular dystrophy.
In Life Science Alliance on 1 January 2025 by Liu, Y., Li, S., et al.
Duchenne muscular dystrophy (DMD) is a lethal disease caused by mutations in the DMD gene that encodes dystrophin. Dystrophin deficiency also impacts muscle stem cells (MuSCs), resulting in impaired asymmetric stem cell division and myogenic commitment. Using MuSCs from DMD patients and the DMD mouse model mdx, we found that PTPN1 phosphatase expression is up-regulated and STAT3 phosphorylation is concomitantly down-regulated in DMD MuSCs. To restore STAT3-mediated myogenic signaling, we examined the effect of K884, a novel PTPN1/2 inhibitor, on DMD MuSCs. Treatment with K884 enhanced STAT3 phosphorylation and promoted myogenic differentiation of DMD patient-derived MuSCs. In MuSCs from mdx mice, K884 treatment increased the number of asymmetric cell divisions, correlating with enhanced myogenic differentiation. Interestingly, the pro-myogenic effect of K884 is specific to human and murine DMD MuSCs and is absent from control MuSCs. Moreover, PTPN1/2 loss-of-function experiments indicate that the pro-myogenic impact of K884 is mediated mainly through PTPN1. We propose that PTPN1/2 inhibition may serve as a therapeutic strategy to restore the myogenic function of MuSCs in DMD.
© 2024 Liu et al.
-
Stem Cells and Developmental Biology
Virus replication is not required for oncolytic bovine herpesvirus-1 immunotherapy.
In Molecular Therapy. Oncology on 19 December 2024 by Baracuhy, E. M., Cormier, O., et al.
Oncolytic viruses are a promising approach for cancer treatment where viruses selectively target and kill cancer cells while also stimulating an immune response. Among viruses with this ability, bovine herpesvirus-1 (BoHV-1) has several advantages, including observations suggesting it may not require viral replication for its anti-cancer effects. We previously demonstrated that binding and penetration of enveloped virus particles are sufficient to trigger intrinsic and innate immune signaling in normal cells, while other groups have published the efficacy of non-replicating viruses as viable immunotherapies in different cancer models. In this work, we definitively show that live and UV-inactivated (UV) (non-replicating) BoHV-1-based regimens extend survival of tumor-bearing mice to similar degrees and induce infiltration of similar immune cell populations, with the exception of neutrophils. Transcriptomic analysis of tumors treated with either live or UV BoHV-1-based regimens revealed similar pathway enrichment and a subset of overlapping differentially regulated genes, suggesting live and UV BoHV-1 have similar mechanisms of activity. Last, we present a gene signature across our in vitro and in vivo models that could potentially be used to validate new BoHV-1 therapeutics. This work contributes to the growing body of literature showing that replication may not be necessary for therapeutic efficacy of viral immunotherapies.
© 2024 The Author(s).
-
Immunology and Microbiology
-
Veterinary Research
Deletion of Gba in neurons, but not microglia, causes neurodegeneration in a Gaucher mouse model.
In JCI Insight on 8 November 2024 by Duffy, H. B., Byrnes, C., et al.
Gaucher disease, the most prevalent lysosomal storage disease, is caused by homozygous mutations at the GBA gene, which is responsible for encoding the enzyme glucocerebrosidase. Neuronopathic Gaucher disease is associated with microgliosis, astrogliosis, and neurodegeneration. However, the role that microglia, astrocytes, and neurons play in the disease remains to be determined. In the current study, we developed inducible, cell-type-specific Gba-KO mice to better understand the individual impacts of Gba deficiencies on microglia and neurons. Gba was conditionally knocked out either exclusively in microglia or neurons or throughout the body. These mouse models were developed using a tamoxifen-inducible Cre system, with tamoxifen administration commencing at weaning. Microglia-specific Gba-KO mice showed no signs of disease. However, the neuron-specific Gba KO resulted in a shortened lifespan, severe weight loss, and ataxia. These mice also had significant neurodegeneration, microgliosis, and astrogliosis accompanied by the accumulation of glucosylceramide and glucosylsphingosine, recapitulating Gaucher disease-like symptoms. These surprising findings reveal that, unlike the neuron-specific Gba deficiency, microglia-specific Gba deficiency alone does not induce disease. The neuronal Gaucher disease mouse model, with a median survival of 16 weeks, may be useful for future studies of pathogenesis and the evaluation of therapies.
-
FC/FACS
-
Mus musculus (House mouse)
-
Neuroscience
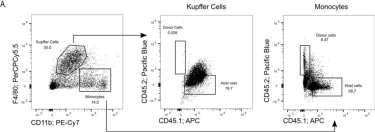
In Int J Mol Sci on 28 February 2022 by Arsiwala, T., Vogt, A. S., et al.
Fig.2.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 7
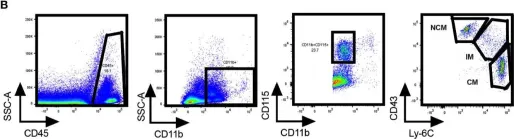
In Int J Mol Sci on 28 February 2022 by Arsiwala, T., Vogt, A. S., et al.
Fig.3.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 7
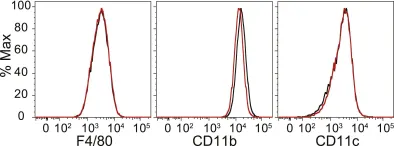
In Int J Mol Sci on 28 February 2022 by Arsiwala, T., Vogt, A. S., et al.
Fig.3.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 7
In Front Oncol on 16 October 2021 by de Almeida, L. Y., Pereira-Martins, D. A., et al.
Fig.3.D

-
FC/FACS
-
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 7
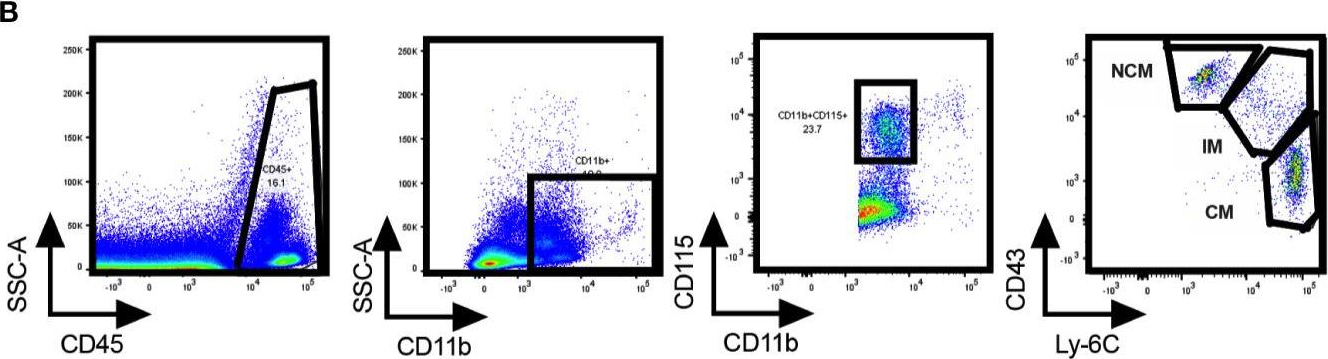
In Front Immunol on 17 November 2020 by Consiglio, C. R. & Gollnick, S. O.
Fig.2.B
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Front Immunol by CiteAb, provided under a CC-BY license
Image 1 of 7
In Mediators Inflamm on 5 April 2019 by Konrad, F. M., Wohlert, J., et al.
Fig.2.A

-
FC/FACS
-
Collected and cropped from Mediators Inflamm by CiteAb, provided under a CC-BY license
Image 1 of 7
In Elife on 30 October 2015 by Freedman, T. S., Tan, Y. X., et al.
Fig.1.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 7