
Oncogenic mutations are widespread in normal human tissues1. Similarly, in murine chimeras, cells carrying an oncogenic lesion contribute normal cells to adult tissues without causing cancer2-4. How lineages that escape cancer via normal development differ from the minority that succumb is unclear. Tumours exhibit characteristic cancer hallmarks; we therefore searched for hallmarks that differentiate cancer-prone lineages from resistant lineages. Here we show that total cell cycle duration (Tc) predicts transformation susceptibility across multiple tumour types. Cancer-prone Rb- and p107-deficient retina (Rb is also known as Rb1 and p107 is also known as Rbl1) exhibited defects in apoptosis, senescence, immune surveillance, angiogenesis, DNA repair, polarity and proliferation. Perturbing the SKP2-p27-CDK2/CDK1 axis could block cancer without affecting these hallmarks. Thus, cancer requires more than the presence of its hallmarks. Notably, every tumour-suppressive mutation that we tested increased Tc, and the Tc of the cell of origin of retinoblastoma cells was half that of resistant lineages. Tc also differentiated the cell of origin in Rb-/- pituitary cancer. In lung, loss of Rb and p53 (also known as Trp53) transforms neuroendocrine cells, whereas KrasG12D or BrafV600E mutations transform alveolar type 2 cells5-7. The shortest Tc consistently identified the cell of origin, regardless of mutation timing. Thus, relative Tc is a hallmark of initiation that distinguishes cancer-prone from cancer-resistant lineages in several settings, explaining how mutated cells escape transformation without inducing apoptosis, senescence or immune surveillance.
© 2025. The Author(s).
Product Citations: 52
Cell cycle duration determines oncogenic transformation capacity.
In Nature on 1 May 2025 by Chen, D., Lu, S., et al.
SOX10 mediates glioblastoma cell-state plasticity.
In EMBO Reports on 1 November 2024 by Man, K. H., Wu, Y., et al.
Phenotypic plasticity is a cause of glioblastoma therapy failure. We previously showed that suppressing the oligodendrocyte-lineage regulator SOX10 promotes glioblastoma progression. Here, we analyze SOX10-mediated phenotypic plasticity and exploit it for glioblastoma therapy design. We show that low SOX10 expression is linked to neural stem-cell (NSC)-like glioblastoma cell states and is a consequence of temozolomide treatment in animal and cell line models. Single-cell transcriptome profiling of Sox10-KD tumors indicates that Sox10 suppression is sufficient to induce tumor progression to an aggressive NSC/developmental-like phenotype, including a quiescent NSC-like cell population. The quiescent NSC state is induced by temozolomide and Sox10-KD and reduced by Notch pathway inhibition in cell line models. Combination treatment using Notch and HDAC/PI3K inhibitors extends the survival of mice carrying Sox10-KD tumors, validating our experimental therapy approach. In summary, SOX10 suppression mediates glioblastoma progression through NSC/developmental cell-state transition, including the induction of a targetable quiescent NSC state. This work provides a rationale for the design of tumor therapies based on single-cell phenotypic plasticity analysis.
© 2024. The Author(s).
The OTUD6B-LIN28B-MYC axis determines the proliferative state in multiple myeloma.
In The EMBO Journal on 17 October 2022 by Paulmann, C., Spallek, R., et al.
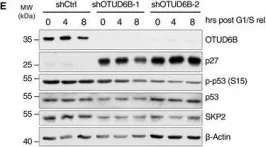
Deubiquitylases (DUBs) are therapeutically amenable components of the ubiquitin machinery that stabilize substrate proteins. Their inhibition can destabilize oncoproteins that may otherwise be undruggable. Here, we screened for DUB vulnerabilities in multiple myeloma, an incurable malignancy with dependency on the ubiquitin proteasome system and identified OTUD6B as an oncogene that drives the G1/S-transition. LIN28B, a suppressor of microRNA biogenesis, is specified as a bona fide cell cycle-specific substrate of OTUD6B. Stabilization of LIN28B drives MYC expression at G1/S, which in turn allows for rapid S-phase entry. Silencing OTUD6B or LIN28B inhibits multiple myeloma outgrowth in vivo and high OTUD6B expression evolves in patients that progress to symptomatic multiple myeloma and results in an adverse outcome of the disease. Thus, we link proteolytic ubiquitylation with post-transcriptional regulation and nominate OTUD6B as a potential mediator of the MGUS-multiple myeloma transition, a central regulator of MYC, and an actionable vulnerability in multiple myeloma and other tumors with an activated OTUD6B-LIN28B axis.
© 2022 The Authors. Published under the terms of the CC BY 4.0 license.
-
WB
In American Journal of Cancer Research on 13 April 2022 by Chung, Y. M., Tsai, W. B., et al.
Boosting anticancer immunity by blocking immune checkpoints such as the programmed death-1 (PD-1) or its ligand (PD-L1) is a breakthrough anticancer therapy. However, many cancer patients do not respond well to immune checkpoint blockades (ICBs) alone. Here we show that low-dose pharmacological immunoactivators (e.g., SN38, topotecan, sorafenib, etc.) notably downregulate PD-L1 and upregulate FOXO3 expression in various human and murine cancer cell lines. In a mouse tumor model, low-dose SN38 treatment markedly suppresses tumor growth, reduces PD-L1 expression, and enhances FOXO3 expression in primary tumor specimens. SN38 therapy engages the tumor-infiltrating mouse NK1.1/CD49b/NKG2D-positive natural killer (NK) cells to attack tumor cells by inducing mouse IFN-γ and granzyme-B secretion in the tumor microenvironment (TME) in vivo. SN38 treatment also promotes tumor cell apoptosis in the TME. SN38 treatment significantly decreases STAT3-pY705 and IL-6 protein levels; FOXO3 is essential for SN38-mediated PD-L1 downregulation. Collectively, these findings may contribute to future translational or clinical investigations tackling difficult-to-treat cancers with immune-activating medicines or combined with ICB immunotherapy.
AJCR Copyright © 2022.
-
Cancer Research
-
Immunology and Microbiology
In Cancers on 4 January 2022 by Nakajima, W., Miyazaki, K., et al.
Epigenetic alterations caused by aberrant DNA methylation have a crucial role in cancer development, and the DNA-demethylating agent decitabine, is used to treat hematopoietic malignancy. Triple-negative breast cancers (TNBCs) have shown sensitivity to decitabine; however, the underlying mechanism of its anticancer effect and its effectiveness in treating TNBCs are not fully understood. We analyzed the effects of decitabine on nine TNBC cell lines and examined genes associated with its cytotoxic effects. According to the effect of decitabine, we classified the cell lines into cell death (D)-type, growth inhibition (G)-type, and resistant (R)-type. In D-type cells, decitabine induced the expression of apoptotic regulators and, among them, NOXA was functionally involved in decitabine-induced apoptosis. In G-type cells, induction of the cyclin-dependent kinase inhibitor, p21, and cell cycle arrest were observed. Furthermore, decitabine enhanced the cytotoxic effect of cisplatin mediated by NOXA in D-type and G-type cells. In contrast, the sensitivity to cisplatin was high in R-type cells, and no enhancing effect by decitabine was observed. These results indicate that decitabine enhances the proapoptotic effect of cisplatin on TNBC cell lines that are less sensitive to cisplatin, indicating the potential for combination therapy in TNBC.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
-
Genetics
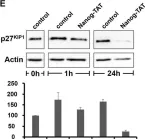
In EMBO J on 17 October 2022 by Paulmann, C., Spallek, R., et al.
Fig.1.E

-
WB
-
Collected and cropped from EMBO J by CiteAb, provided under a CC-BY license
Image 1 of 9
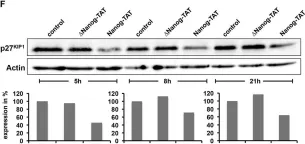
In Cancers (Basel) on 4 January 2022 by Nakajima, W., Miyazaki, K., et al.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 9
In PLoS One on 29 November 2017 by Mayoral-Varo, V., Calcabrini, A., et al.
Fig.2.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 9
In Nat Commun on 16 January 2017 by Xu, J., Zhou, W., et al.
Fig.3.H

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 9
In Nat Commun on 16 January 2017 by Xu, J., Zhou, W., et al.
Fig.5.G

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Cell Sci on 1 March 2016 by Münst, B., Thier, M. C., et al.
Fig.2.F

-
WB
-
Collected and cropped from J Cell Sci by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Cell Sci on 1 March 2016 by Münst, B., Thier, M. C., et al.
Fig.2.E

-
WB
-
Collected and cropped from J Cell Sci by CiteAb, provided under a CC-BY license
Image 1 of 9
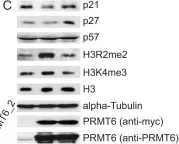
In PLoS One on 24 August 2012 by Kleinschmidt, M. A., de Graaf, P., et al.
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 9
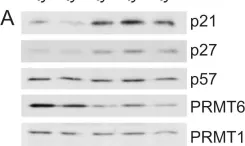
In PLoS One on 24 August 2012 by Kleinschmidt, M. A., de Graaf, P., et al.
Fig.1.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 9