
Cellular quiescence is a state of reversible proliferative arrest that plays essential roles in development, resistance to stress, aging, and longevity of organisms. Here we report that rapid depletion of RNase MRP, a deeply conserved RNA-based enzyme required for rRNA biosynthesis, induces a long-term yet reversible proliferative arrest in human cells. Severely compromised biogenesis of rRNAs along with acute transcriptional reprogramming precede a gradual decline of the critical cellular functions. Unexpectedly, many arresting cells show increased levels of histone mRNAs, which accumulate locally in the cytoplasm, and S-phase DNA amount. The ensuing proliferative arrest is entered from multiple stages of the cell cycle and can last for several weeks with uncompromised cell viability. Strikingly, restoring expression of RNase MRP leads to a complete reversal of the arrested state with resumed cell proliferation at the speed of control cells. We suggest that targeting rRNA biogenesis may provide a general strategy for rapid induction of a reversible proliferative arrest, with implications for understanding and manipulating cellular quiescence.
© 2025. The Author(s).
Product Citations: 608
Reversible proliferative arrest induced by rapid depletion of RNase MRP.
In Nature Communications on 18 June 2025 by Liu, Y., He, S., et al.
In Tumour Virus Research on 1 June 2025 by Gelbard, M. K., Grace, M., et al.
Human papillomaviruses (HPVs) are a diverse family of viruses with over 450 members that have been identified and fully sequenced. They are classified into five phylogenetic genera: alpha, beta, gamma, mu, and nu. The high-risk alpha HPVs, such as HPV16, have been studied the most extensively due to their medical significance as cancer-causing agents. However, while nearly 70% of all HPVs are members of the gamma genus, they are almost entirely unstudied. This is because gamma HPVs have been considered medically irrelevant commensals as most of them infect the skin and are not known to cause significant clinical lesions in immunocompetent individuals. Members of the gamma 6 HPVs, however, have been detected in the anogenital tract mucosa and HPV101 has been isolated from a premalignant cervical lesion. Moreover, gamma 6 HPVs have a unique genome structure. They lack E6 proteins but in place of E6, they encode unique, small hydrophobic proteins without any close viral or cellular homologs that have been termed E10. Here, we report that HPV101 E7 shares biochemical activities with the high-risk alpha HPV16 E7, including the ability to target the pRB and PTPN14 tumor suppressors for degradation. This study underscores the importance of further characterizing HPV101 and other unstudied HPV species.
Copyright © 2024 The Authors. Published by Elsevier B.V. All rights reserved.
Loss of VHL-mediated pRb regulation promotes clear cell renal cell carcinoma.
In Cell Death & Disease on 16 April 2025 by Akuma, M., Kim, M., et al.
The von Hippel-Lindau (VHL) tumor suppressor is a substrate-defining component of E3 ubiquitin ligase complexes that target cellular substrates for proteasome-mediated degradation. VHL inactivation by mutation or transcriptional silencing is observed in most sporadic cases of clear cell renal cell carcinoma (ccRCC). VHL loss in ccRCC leads to constitutive stabilization of E3 ligase substrates, including hypoxia inducible factor α (HIFα). HIFα stabilization upon VHL loss is known to contribute to ccRCC development through transactivation of hypoxia-responsive genes. HIF-independent VHL targets have been implicated in oncogenesis, although those mechanisms are less well-defined than for HIFα. Using proximity labeling to identify proteasomal-sensitive VHL interactors, we identified retinoblastoma protein (pRb) as a novel substrate of VHL. Mechanistically, VHL interacts with pRb in a proteasomal-sensitive manner, promoting its ubiquitin-mediated degradation. Concordantly, VHL-inactivation results in pRb hyperstabilization. Functionally, loss of pRb in ccRCC led to increased cell death, transcriptional changes, and loss of oncogenic properties in vitro and in vivo. We also show that downstream transcriptional changes induced by pRb hyperstabilization may contribute to ccRCC tumor development. Together, our findings reveal a novel VHL-related pathway which can be therapeutically targeted to inhibit ccRCC tumor development.
© 2025. The Author(s).
-
WB
-
Cancer Research
-
Cell Biology
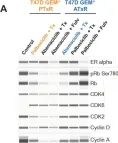
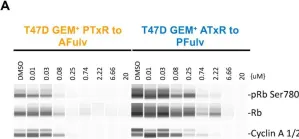
Models of Early Resistance to CDK4/6 Inhibitors Unveil Potential Therapeutic Treatment Sequencing.
In International Journal of Molecular Sciences on 14 March 2025 by Zapatero-Solana, E., Ding, Y., et al.
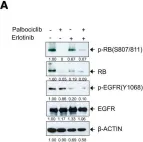
CDK4/6 inhibitors (CDK4/6i) combined with hormone therapies have demonstrated clinical benefit in HR+, HER2- breast cancer patients. However, the onset of resistance remains a concern and highlights a need for therapeutic strategies to improve outcomes. The objective of this study was to develop an in vitro model to better understand the mechanisms of resistance to CDK4/6i + hormone therapies and identify therapeutic strategies with potential to overcome this resistance.
The HR+, HER2- T47D breast cancer cell line genetically modified with a Geminin-Venus reporter construct was treated with CDK4/6i (abemaciclib or palbociclib) in combination with 4-hydroxytamoxifen (tamoxifen). Resistant cells were identified by cell sorting for Geminin (%GEM+), a marker of the S/G2/M phases of the cell cycle, and confirmed by treatment with tamoxifen plus the CDK4/6i used to drive resistance. In resistant cells, following treatment with CDK4/6i + ET (tamoxifen or fulvestrant), the effects on cell proliferation (%GEM+) and viability, gene expression, and protein analysis to evaluate CDK4/6-cyclin D complex composition were examined.
Palbociclib + tamoxifen-resistant (PTxR) cells treated with abemaciclib + ET showed decreased %GEM+, %Ki67, and colony formation ability, compared to abemaciclib + tamoxifen-resistant (ATxR) cells treated with palbociclib + ET. Additionally, PTxR cells showed increased CDK4-p21 interaction, compared to ATxR. The CDK6 levels were greater in ATxR cells compared to PTxR cells, associated with CDK4/6i resistance. Additionally, abemaciclib + fulvestrant continued to robustly decrease pRb levels in PTxR models compared to palbociclib + fulvestrant in ATxR models. Transcriptome analysis revealed a depression of the cell cycle and E2F- and Rb-related genes in PTxR cells following treatment with abemaciclib + ET, not present in ATxR cells treated with palbociclib + ET. Both resistant models showed increased EGFR-related gene expression.
Taken together, we describe CDK4/6i-dependent mechanisms resulting in early-onset resistance to CDK4/6i + ET, using clinically relevant drug concentrations, in preclinical breast cancer cell models. The characterization of these preclinical models post progression on CDK4/6 inhibitor + ET treatment highlights the potential that the specific sequencing of CDK4/6 inhibitors could offer to overcome acquired resistance to CDK4/6i + ET. Abemaciclib + fulvestrant is currently under clinical investigation in patients with HR+, HER2- breast cancer and progression on prior CDK4/6i + ET (NCT05169567, postMONARCH).
-
WB
In Journal of Virology on 22 October 2024 by Schaal, D. L., Amucheazi, A. A., et al.
Epstein-Barr virus (EBV) co-infections with human papillomavirus (HPV) have been observed in oropharyngeal squamous cell carcinoma. Modeling EBV/HPV co-infection in organotypic epithelial raft cultures revealed that HPV16 E7 inhibited EBV productive replication through the facilitated degradation of the retinoblastoma protein pRb/p105. To further understand how pRb is required for EBV productive replication, we generated CRISPR-Cas9 pRb knockout (KO) normal oral keratinocytes (NOKs) in the context of wild-type and mutant K120E p53. EBV replication was examined in organotypic rafts as a physiological correlate for epithelial differentiation. In pRb KO rafts, EBV DNA copy number was statistically decreased compared to vector controls, regardless of p53 context. Loss of pRb did not affect EBV binding or internalization of calcium-treated NOKs or early infection of rafts. Rather, the block in EBV replication correlated with impaired immediate early gene expression. An EBV infection time course in rafts with mutant p53 demonstrated that pRb-positive basal cells were initially infected with delayed replication occurring in differentiated layers. Loss of pRb showed increased S-phase progression makers and elevated activator E2F activity in raft tissues. Complementation with a panel of pRb/E2F binding mutants showed that wild type or pRb∆685 mutant capable of E2F binding reduced S-phase marker gene expression, rescued EBV DNA replication, and restored BZLF1 expression in pRb KO rafts. However, pRb KO complemented with pRb661W mutant, unable to bind E2Fs, failed to rescue EBV replication in raft culture. These findings suggest that EBV productive replication in differentiated epithelium requires pRb inhibition of activator E2Fs to restrict S-phase progression.IMPORTANCEA subset of human papillomavirus (HPV)-positive oropharyngeal squamous cell carcinoma is co-positive for Epstein-Barr virus (EBV). Potential oncogenic viral interactions revealed that HPV16 E7 inhibited productive EBV replication within the differentiated epithelium. As E7 mediates the degradation of pRb, we aimed to establish how pRb is involved in EBV replication. In the context of differentiated epithelium using organotypic raft culture, we evaluated how the loss of pRb affects EBV lytic replication to better comprehend EBV contributions to carcinogenesis. In this study, ablation of pRb interfered with EBV replication at the level of immediate early gene expression. Loss of pRb increased activator E2Fs and associated S-phase gene expression throughout the differentiated epithelium. Complementation studies showed that wild type and pRb mutant capable of binding to E2F rescued EBV replication, while pRb mutant lacking E2F binding did not. Altogether, these studies support that in differentiated tissues, HPV16 E7-mediated degradation of pRb inhibits EBV replication through unregulated E2F activity.
-
WB
-
Biochemistry and Molecular biology
-
Immunology and Microbiology
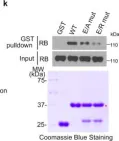
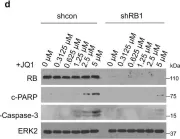
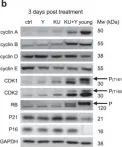
In Int J Mol Sci on 14 March 2025 by Zapatero-Solana, E., Ding, Y., et al.
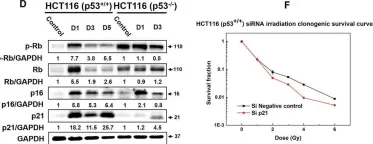
Fig.5.A

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 56
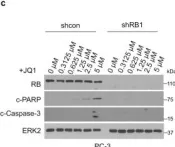
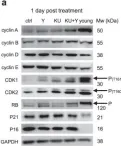
In Int J Mol Sci on 14 March 2025 by Zapatero-Solana, E., Ding, Y., et al.
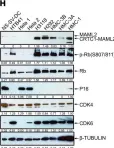
Fig.2.A

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 56
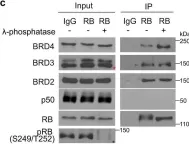
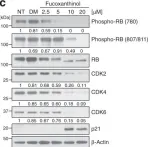
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.3.H

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
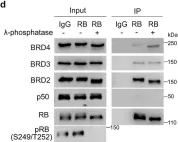
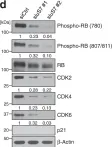
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.6.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
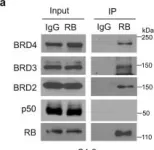
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.3.K

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.2.C

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.2.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.2.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.1.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.1.C

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 23 October 2022 by Ding, D., Zheng, R., et al.
Fig.2.B

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Commun Biol on 14 July 2022 by Yang, E. J., Park, J. H., et al.
Fig.5.D

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
In Commun Biol on 14 July 2022 by Yang, E. J., Park, J. H., et al.
Fig.5.B

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
In Commun Biol on 14 July 2022 by Yang, E. J., Park, J. H., et al.
Fig.5.A

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
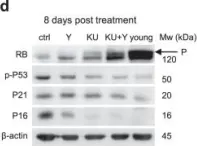
In Commun Biol on 9 June 2022 by Iizumi, Y., Sowa, Y., et al.
Fig.1.C

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
In Commun Biol on 9 June 2022 by Iizumi, Y., Sowa, Y., et al.
Fig.4.D

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cell Death Discov on 20 July 2021 by Li, P., Liu, X., et al.
Fig.3.D

-
WB
-
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 56
In JCI Insight on 8 April 2021 by Chen, Z., Ni, W., et al.
Fig.4.H

-
WB
-
Collected and cropped from JCI Insight by CiteAb, provided under a CC-BY license
Image 1 of 56
In JCI Insight on 8 April 2021 by Chen, Z., Ni, W., et al.
Fig.5.A

-
WB
-
Collected and cropped from JCI Insight by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 7 April 2021 by Dewhurst, S. M., Yao, X., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
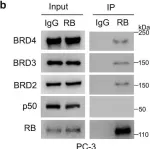
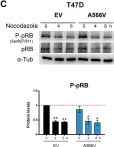
In Theranostics on 5 February 2021 by Verdugo-Sivianes, E. M., Rojas, A. M., et al.
Fig.7.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Theranostics by CiteAb, provided under a CC-BY license
Image 1 of 56
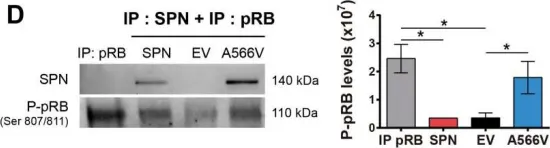
In Theranostics on 5 February 2021 by Verdugo-Sivianes, E. M., Rojas, A. M., et al.
Fig.5.D

-
IP
-
Homo sapiens (Human)
Collected and cropped from Theranostics by CiteAb, provided under a CC-BY license
Image 1 of 56
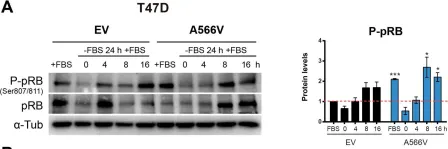
In Theranostics on 5 February 2021 by Verdugo-Sivianes, E. M., Rojas, A. M., et al.
Fig.6.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Theranostics by CiteAb, provided under a CC-BY license
Image 1 of 56
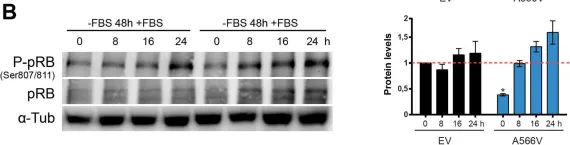
In Theranostics on 5 February 2021 by Verdugo-Sivianes, E. M., Rojas, A. M., et al.
Fig.7.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Theranostics by CiteAb, provided under a CC-BY license
Image 1 of 56
In Theranostics on 5 February 2021 by Verdugo-Sivianes, E. M., Rojas, A. M., et al.
Fig.6.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Theranostics by CiteAb, provided under a CC-BY license
Image 1 of 56