

Human papillomaviruses (HPV) infect epithelia to cause benign lesions or warts. However, the so-called "high risk" HPVs infecting the anogenital region and the oropharynx can cause precancerous lesions that may progress to malignant tumours. Understanding the HPV life cycle is important to the discovery of novel antiviral therapies. HPV uses cellular splicing to produce the full suite of viral mRNAs. Members of the serine/arginine rich (SR) protein family can positively regulate splicing. SR protein activity and cellular location is regulated by phosphorylation of their serine-arginine domains. SR protein kinases (SRPK) can phosphorylate SR proteins. This licenses their nuclear entry and promotes nuclear splicing together with another SR protein kinase, Clk1. SRPIN340 is a specific inhibitor of SRPK1. It has been reported to inhibit replication of HCV, Sindbis virus and HIV. We show here that SRPIN340 inhibits the expression of the viral replication and transcription factor, E2. Loss of HPV E4 and L1 late proteins was also observed. RNA sequencing showed SRPIN340 treatment resulted in gene expression changes opposite to those induced by HPV16 infection. In particular, the loss of the epithelial barrier was restored. SRPIN340 treatment led to changes in alternative splicing of 935 RNAs and pathway analysis showed a predominance of changes to RNAs encoding proteins involved in chromatin conformation, DNA repair and RNA processing. Short term SRPIN340 treatment (two to three days) was not associated with changes in proliferation or differentiation of keratinocytes. Since SRPK1 controls the E2 viral replication and transcription factor, targeting this kinase, or the phosphorylation events it mediates, could be considered as a therapeutic strategy for HPV16 infection.
Copyright: © 2025 Faizo et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Product Citations: 49
In PLoS Pathogens on 1 April 2025 by Faizo, A. A. A., Bellward, C., et al.
-
WB
-
Immunology and Microbiology
Preprint on BioRxiv : the Preprint Server for Biology on 28 October 2024 by Faizo, A. A. A., Bellward, C., et al.
ABSTRACT Human papillomaviruses (HPV) infect epithelial causing benign lesions. However, the so-called “high risk” HPVs that infect the anogenital regions and the oropharynx can cause precancerous lesions that can progress to malignant tumours. Understanding the HPV life cycle is important to the discovery of novel antiviral therapies. HPV uses cellular splicing for the production of the full suite of viral mRNAs. Members of the serine/arginine rich (SR) protein family can positively regulate splicing. SR protein activity and cellular location is regulated by kinases through phosphorylation of their serine-arginine domains. SR protein kinase 1 (SRPK1) phosphorylates SR proteins to licence their nuclear entry and can promote nuclear splicing in conjunction with another SR protein kinase, Clk1. SRPIN340 is a specific SRPK1 inhibitor that has been reported to inhibit replication of HCV, Sindbis virus and HIV. Using organotypic raft cultures supporting the HPV16 life cycle, we show that SRPIN340 inhibits the expression of the viral replication and transcription factor, E2. Drug-mediated loss of E2 is specifically associated with inhibition of late gene expression since early gene expression is unaffected. RNA sequencing revealed that SRPIN340 treatment resulted in many gene expression changes opposite to those induced by HPV16 infection. In particular, the loss of epithelial barrier structure and immune function was restored. SRPIN340 treatment resulted in changes in alternative splicing of 935 RNAs and pathway analysis showed a predominance of changes to RNAs encoding proteins involved in chromatin conformation, DNA repair and RNA processing. SRPIN340 treatment was not associated with changes in proliferation or differentiation of keratinocytes. Collectively, these results provide evidence that SRPK1 is a host factor that is essential for HPV16 replication and therefore, targeting this factor, or the phosphorylation events it mediates, could be considered as a therapeutic strategy for HPV16 infection. AUTHOR SUMMARY Human papillomavirus (HPV) can cause precancerous and cancerous tumours of the anogenital region, for example, the cervix. HPV infects epithelial cells and utilizes the host cell gene expression machinery to produce its own proteins. Splicing is a key mechanism used by HPV to synthesise messenger RNAs that encode the viral proteins. Splicing is positively controlled by SR proteins which require to be phosphorylated by SR protein kinase (SRPK) to enter the nucleus and regulate splicing. SRPIN340 is a specific inhibitor of SRPK. SRPIN340 treatment of HPV16-infected epithelial cells resulted in loss of expression of the viral replication and transcription factor, E2 and abrogation of late events in the viral life cycle. SRPIN340 treatment counteracted some gene expression changes to the epithelium known to be cause by HPV infection. These data suggest that SRPK1 is essential for the HPV life cycle and that it could be a potential anti-viral target.
In Journal of Neuroinflammation on 12 April 2022 by Vicente-Acosta, A., Gimenez-Cassina, A., et al.
Friedreich's ataxia is a rare hereditary neurodegenerative disease caused by decreased levels of the mitochondrial protein frataxin. Similar to other neurodegenerative pathologies, previous studies suggested that astrocytes might contribute to the progression of the disease. To fully understand the mechanisms underlying neurodegeneration in Friedreich's ataxia, we investigated the reactivity status and functioning of cultured human astrocytes after frataxin depletion using an RNA interference-based approach and tested the effect of pharmacologically modulating the SHH pathway as a novel neuroprotective strategy.
We observed loss of cell viability, mitochondrial alterations, increased autophagy and lipid accumulation in cultured astrocytes upon frataxin depletion. Besides, frataxin-deficient cells show higher expression of several A1-reactivity markers and release of pro-inflammatory cytokines. Interestingly, most of these defects were prevented by chronically treating the cells with the smoothened agonist SAG. Furthermore, in vitro culture of neurons with conditioned medium from frataxin-deficient astrocytes results in a reduction of neuronal survival, neurite length and synapse formation. However, when frataxin-deficient astrocytes were chronically treated with SAG, we did not observe these alterations in neurons.
Our results demonstrate that the pharmacological activation of the SHH pathway could be used as a target to modulate astrocyte reactivity and neuron-glia interactions to prevent neurodegeneration in Friedreich's ataxia.
© 2022. The Author(s).
-
WB
-
Cell Biology
-
Immunology and Microbiology
-
Neuroscience
In Nature Medicine on 1 March 2022 by Ganan-Gomez, I., Yang, H., et al.
Myelodysplastic syndromes (MDS) are heterogeneous neoplastic disorders of hematopoietic stem cells (HSCs). The current standard of care for patients with MDS is hypomethylating agent (HMA)-based therapy; however, almost 50% of MDS patients fail HMA therapy and progress to acute myeloid leukemia, facing a dismal prognosis due to lack of approved second-line treatment options. As cancer stem cells are the seeds of disease progression, we investigated the biological properties of the MDS HSCs that drive disease evolution, seeking to uncover vulnerabilities that could be therapeutically exploited. Through integrative molecular profiling of HSCs and progenitor cells in large patient cohorts, we found that MDS HSCs in two distinct differentiation states are maintained throughout the clinical course of the disease, and expand at progression, depending on recurrent activation of the anti-apoptotic regulator BCL-2 or nuclear factor-kappa B-mediated survival pathways. Pharmacologically inhibiting these pathways depleted MDS HSCs and reduced tumor burden in experimental systems. Further, patients with MDS who progressed after failure to frontline HMA therapy and whose HSCs upregulated BCL-2 achieved improved clinical responses to venetoclax-based therapy in the clinical setting. Overall, our study uncovers that HSC architectures in MDS are potential predictive biomarkers to guide second-line treatments after HMA failure. These findings warrant further investigation of HSC-specific survival pathways to identify new therapeutic targets of clinical potential in MDS.
© 2022. The Author(s).
-
CyTOF
-
Homo sapiens (Human)
-
Stem Cells and Developmental Biology
In Molecular and Cellular Biology on 26 October 2021 by Dall'Osto, M., Pierini, L., et al.
DNA polymerase kappa (Pol κ) has been well documented thus far for its specialized DNA synthesis activity during translesion replication, progression of replication forks through regions difficult to replicate, restart of stalled forks, and replication checkpoint efficiency. Pol κ is also required for the stabilization of stalled forks, although the mechanisms are poorly understood. In this study, we unveiled an unexpected role for Pol κ in controlling the stability and abundance of checkpoint kinase 1 (Chk1), an important actor for the replication checkpoint and fork stabilization. We found that loss of Pol κ decreased the Chk1 protein level in the nuclei of four human cell lines. Pol κ and not the other Y family polymerase members is required to maintain the Chk1 protein pool all along the cell cycle. We showed that Pol κ depletion affected the protein stability of Chk1 and protected it from proteasome degradation. Importantly, we also observed that the fork restart defects observed in Pol κ-depleted cells could be overcome by the reexpression of Chk1. Strikingly, this new function of Pol κ does not require its catalytic activity. We propose that Pol κ could contribute to the protection of stalled forks through Chk1 stability.
-
Homo sapiens (Human)
-
Cell Biology
-
Genetics
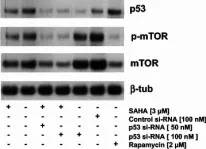
In PLoS One on 2 September 2017 by Sotillo, W. S., Villagomez, R., et al.
Fig.5.H

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
In PLoS One on 2 September 2017 by Sotillo, W. S., Villagomez, R., et al.
Fig.5.G

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
In Cancer Cell Int on 8 September 2016 by Fröhlich, L. F., Mrakovcic, M., et al.
Fig.7.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cancer Cell Int by CiteAb, provided under a CC-BY license
Image 1 of 13
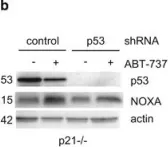
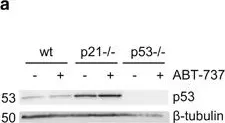
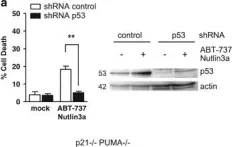
In Cell Death Dis on 4 February 2016 by Le Pen, J., Maillet, L., et al.
Fig.2.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 13
In Cell Death Dis on 4 February 2016 by Le Pen, J., Maillet, L., et al.
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 13
In Cell Death Dis on 4 February 2016 by Le Pen, J., Maillet, L., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 13
In Cell Death Dis on 4 February 2016 by Le Pen, J., Maillet, L., et al.
Fig.1.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 13
In Cell Death Dis on 4 February 2016 by Le Pen, J., Maillet, L., et al.
Fig.2.C

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 13
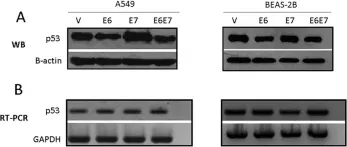
In PLoS One on 5 June 2012 by Muñoz, J. P., Gonzalez, C., et al.
Fig.2.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
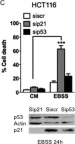
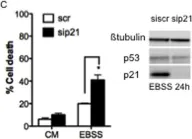
In PLoS One on 3 September 2011 by Braun, F., Bertin-Ciftci, J., et al.
Fig.1.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
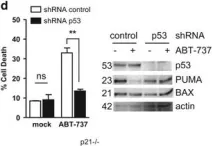
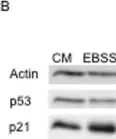
In PLoS One on 3 September 2011 by Braun, F., Bertin-Ciftci, J., et al.
Fig.2.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
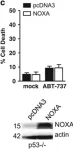
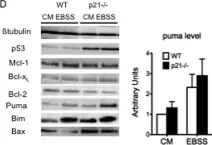
In PLoS One on 3 September 2011 by Braun, F., Bertin-Ciftci, J., et al.
Fig.3.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13
In PLoS One on 3 September 2011 by Braun, F., Bertin-Ciftci, J., et al.
Fig.3.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 13