Immune checkpoint inhibitors (ICI) have revolutionized cancer therapy, but their use is limited by the development of autoimmunity in healthy tissues as a side effect of treatment. Such immune-related adverse events (IrAE) contribute to hospitalizations, cancer treatment interruption, and even premature death. ICI-induced autoimmune diabetes mellitus (ICI-T1DM) is a life-threatening IrAE that presents with rapid pancreatic β-islet cell destruction leading to hyperglycemia and life-long insulin dependence. While prior reports have focused on CD8+ T cells, the role for CD4+ T cells in ICI-T1DM is less understood. We identify expansion of CD4+ T follicular helper (Tfh) cells expressing IL-21 and IFN-γ as a hallmark of ICI-T1DM. Furthermore, we show that both IL-21 and IFN-γ are critical cytokines for autoimmune attack in ICI-T1DM. Because IL-21 and IFN-γ both signal through JAK/STAT pathways, we reasoned that JAK inhibitors (JAKi) may protect against ICI-T1DM. Indeed, JAKi provide robust in vivo protection against ICI-T1DM in a mouse model that is associated with decreased islet-infiltrating Tfh cells. Moreover, JAKi therapy impaired Tfh cell differentiation in patients with ICI-T1DM. These studies highlight CD4+ Tfh cells as underrecognized but critical mediators of ICI-T1DM that may be targeted with JAKi to prevent this grave IrAE.
Product Citations: 10
In JCI Insight on 8 July 2025 by Huang, N. L., Ortega, J. G., et al.
Preprint on BioRxiv : the Preprint Server for Biology on 3 December 2024 by Huang, N., Ortega, J., et al.
ABSTRACT Immune checkpoint inhibitors (ICI) have revolutionized cancer therapy, but their use is limited by the development of autoimmunity in healthy tissues as a side effect of treatment. Such immune-related adverse events (IrAE) contribute to hospitalizations, cancer treatment interruption and even premature death. ICI-induced autoimmune diabetes mellitus (ICI-T1DM) is a life-threatening IrAE that presents with rapid pancreatic beta-islet cell destruction leading to hyperglycemia and life-long insulin dependence. While prior reports have focused on CD8 + T cells, the role for CD4 + T cells in ICI-T1DM is less understood. Here, we identify expansion CD4 + T follicular helper (Tfh) cells expressing interleukin 21 (IL-21) and interferon gamma (IFNγ) as a hallmark of ICI-T1DM. Furthermore, we show that both IL-21 and IFNγ are critical cytokines for autoimmune attack in ICI-T1DM. Because IL-21 and IFNγ both signal through JAK-STAT pathways, we reasoned that JAK inhibitors (JAKi) may protect against ICI-T1DM. Indeed, JAKi provide robust in vivo protection against ICI-T1DM in a mouse model that is associated with decreased islet-infiltrating Tfh cells. Moreover, JAKi therapy impaired Tfh cell differentiation in patients with ICI-T1DM. These studies highlight CD4 + Tfh cells as underrecognized but critical mediators of ICI-T1DM that may be targeted with JAKi to prevent this grave IrAE. VISUAL ABSTRACT
-
Mus musculus (House mouse)
Identification of a novel enhancer essential for Satb1 expression in TH2 cells and activated ILC2s.
In Life Science Alliance on 1 August 2023 by Nomura, A., Kobayashi, T., et al.
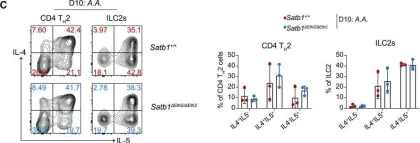
The genome organizer, special AT-rich binding protein-1 (SATB1), functions to globally regulate gene networks during primary T cell development and plays a pivotal role in lineage specification in CD4+ helper-, CD8+ cytotoxic-, and FOXP3+ regulatory-T cell subsets. However, it remains unclear how Satb1 gene expression is controlled, particularly in effector T cell function. Here, by using a novel reporter mouse strain expressing SATB1-Venus and genome editing, we have identified a cis-regulatory enhancer, essential for maintaining Satb1 expression specifically in TH2 cells. This enhancer is occupied by STAT6 and interacts with Satb1 promoters through chromatin looping in TH2 cells. Reduction of Satb1 expression, by the lack of this enhancer, resulted in elevated IL-5 expression in TH2 cells. In addition, we found that Satb1 is induced in activated group 2 innate lymphoid cells (ILC2s) through this enhancer. Collectively, these results provide novel insights into how Satb1 expression is regulated in TH2 cells and ILC2s during type 2 immune responses.
© 2023 Nomura et al.
-
FC/FACS
Preprint on BioRxiv : the Preprint Server for Biology on 3 January 2023 by Nomura, A., Ohno-Oishi, M., et al.
The genome organizer, special AT-rich binding protein-1 (SATB1) functions to globally regulate gene networks during primary T cells development and plays a pivotal role in lineage-specification in CD4 + helper-, CD8 + cytotoxic- and FOXP3 + regulatory-T cell subsets. However, it remains unclear how Satb1 gene expression is controlled, particularly in effector T cell function. Here, by using a novel reporter mouse strain expressing SATB1-Venus and genome editing, we have identified a cis -regulatory enhancer, essential for maintaining Satb1 expression specifically in T H 2 cells. This enhancer is occupied by STAT6 and interacts with Satb1 promoters through chromatin looping in T H 2 cells. Reduction of Satb1 expression, by the lack of this enhancer, resulted in elevated IL-5 expression in T H 2 cells. In addition, we found that Satb1 is induced in activated group 2 innate lymphoid cells (ILC2s) through this enhancer. Collectively, these results provide novel insights into how Satb1 expression is regulated in T H 2 cells and ILC2s during type 2 immune responses.
-
FC/FACS
-
Mus musculus (House mouse)
In The Journal of Clinical Investigation on 1 July 2020 by Wang, Z., Zhao, M., et al.
T follicular helper (Tfh) cells are indispensable for the formation of germinal center (GC) reactions, whereas T follicular regulatory (Tfr) cells inhibit Tfh-mediated GC responses. Aberrant activation of Tfh cells contributes substantially to the pathogenesis of autoimmune diseases, such as systemic lupus erythematosus (SLE). Nonetheless, the molecular mechanisms mitigating excessive Tfh cell differentiation are not fully understood. Herein we demonstrate that the adenovirus E4 promoter-binding protein (E4BP4) mediates a feedback loop and acts as a transcriptional brake to inhibit Tfh cell differentiation. Furthermore, we show that such an immunological mechanism is compromised in patients with SLE. Establishing mice with either conditional knockout (cKO) or knockin (cKI) of the E4bp4 gene in T cells reveals that E4BP4 strongly inhibits Tfh cell differentiation. Mechanistically, E4BP4 regulates Bcl6 transcription by recruiting the repressive epigenetic modifiers HDAC1 and EZH2. E4BP4 phosphorylation site mutants have limited capability with regard to inhibiting Tfh cell differentiation. In SLE, we detected impaired phosphorylation of E4BP4, finding that this compromised transcription factor is positively correlated with disease activity. These findings unveiled molecular mechanisms by which E4BP4 restrains Tfh cell differentiation, whose compromised function is associated with uncontrolled autoimmune reactions in SLE.
-
Immunology and Microbiology
In Life Sci Alliance on 1 August 2023 by Nomura, A., Kobayashi, T., et al.
Fig.5.C

-
FC/FACS
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 1