Multiple sclerosis (MS) is a chronic autoimmune disease of the CNS that is characterized by demyelination, axonal loss, gliosis, and inflammation. The murine model of MS is the experimental autoimmune encephalopathy (EAE) induced by immunization of mice with myelin oligodendrocyte glycoprotein (MOG)35-55 Ig-like transcript 3 (ILT3) is an inhibitory cell surface receptor expressed by tolerogenic human dendritic cells. In this study, we show that the recombinant human ILT3.Fc protein binds to murine immune cells and inhibits the release of proinflammatory cytokines that cause the neuroinflammatory process that result in paralysis. Administration of ILT3.Fc prevents the rapid evolution of the disease in C57BL/6 mice and is associated with a profound reduction of proliferation of MOG35-55-specific Th1 and Th17 cells. Inhibition of IFN-γ and IL-17A in mice treated with ILT3.Fc is associated with delayed time of onset of the disease and its evolution to a peak clinical score. Neuropathological analysis shows a reduction in inflammatory infiltrates and demyelinated areas in the brains and spinal cords of treated mice. These results indicate that inhibition of Th1 and Th17 development provides effective suppression of EAE and suggests the feasibility of a clinical approach based on the use of ILT3.Fc for treatment of MS. Furthermore, our results open the way to further studies on the effect of the human ILT3.Fc protein in murine experimental models of autoimmunity and cancer.
Copyright © 2021 by The American Association of Immunologists, Inc.
Product Citations: 14
Suppression of Experimental Autoimmune Encephalomyelitis by ILT3.Fc.
In The Journal of Immunology on 1 February 2021 by Xu, Z., Lin, C. C., et al.
-
Immunology and Microbiology
Noncanonical STAT3 activity sustains pathogenic Th17 proliferation and cytokine response to antigen.
In The Journal of Experimental Medicine on 5 October 2020 by Poholek, C. H., Raphael, I., et al.
The STAT3 signaling pathway is required for early Th17 cell development, and therapies targeting this pathway are used for autoimmune disease. However, the role of STAT3 in maintaining inflammatory effector Th17 cell function has been unexplored. Th17ΔSTAT3 mice, which delete STAT3 in effector Th17 cells, were resistant to experimental autoimmune encephalomyelitis (EAE), a murine model of MS. Th17 cell numbers declined after STAT3 deletion, corresponding to reduced cell cycle. Th17ΔSTAT3 cells had increased IL-6-mediated phosphorylation of STAT1, known to have antiproliferative functions. Th17ΔSTAT3 cells also had reduced mitochondrial membrane potential, which can regulate intracellular Ca2+. Accordingly, Th17ΔSTAT3 cells had reduced production of proinflammatory cytokines when stimulated with myelin antigen but normal production of cytokines when TCR-induced Ca2+ flux was bypassed with ionomycin. Thus, early transcriptional roles of STAT3 in developing Th17 cells are later complimented by noncanonical STAT3 functions that sustain pathogenic Th17 cell proliferation and cytokine production.
© 2020 Poholek et al.
-
Immunology and Microbiology
In The Journal of Immunology on 15 September 2019 by Sie, C., Perez, L. G., et al.
Homing of pathogenic CD4+ T cells to the CNS is dependent on α4 integrins. However, it is uncertain whether α4 integrins are also required for the migration of dendritic cell (DC) subsets, which sample Ags from nonlymphoid tissues to present it to T cells. In this study, after genetic ablation of Itga4 in DCs and monocytes in mice via the promoters of Cd11c and Lyz2 (also known as LysM), respectively, the recruitment of α4 integrin-deficient conventional and plasmacytoid DCs to the CNS was unaffected, whereas α4 integrin-deficient, monocyte-derived DCs accumulated less efficiently in the CNS during experimental autoimmune encephalomyelitis in a competitive setting than their wild-type counterparts. In a noncompetitive setting, α4 integrin deficiency on monocyte-derived DCs was fully compensated. In contrast, in small intestine and colon, the fraction of α4 integrin-deficient CD11b+CD103+ DCs was selectively reduced in steady-state. Yet, T cell-mediated inflammation and host defense against Citrobacter rodentium were not impaired in the absence of α4 integrins on DCs. Thus, inflammatory conditions can promote an environment that is indifferent to α4 integrin expression by DCs.
Copyright © 2019 by The American Association of Immunologists, Inc.
-
Immunology and Microbiology
NKG2D and Its Ligand MULT1 Contribute to Disease Progression in a Mouse Model of Multiple Sclerosis.
In Frontiers in Immunology on 23 February 2019 by Legroux, L., Moratalla, A. C., et al.
NKG2D is an activating receptor expressed on the surface of immune cells including subsets of T lymphocytes. NKG2D binds multiple ligands (NKG2DL) whose expression are differentially triggered in a cell type and stress specific manner. The NKG2D-NKG2DL interaction has been involved in autoimmune disorders but its role in animal models of multiple sclerosis (MS) remains incompletely resolved. Here we show that NKG2D and its ligand MULT1 contribute to the pathobiology of experimental autoimmune encephalomyelitis (EAE). MULT1 protein levels are increased in the central nervous system (CNS) at EAE disease peak; soluble MULT1 is elevated in the cerebrospinal fluid of both active and passive EAE. We establish that such soluble MULT1 enhances effector functions (e.g., IFNγ production) of activated CD8 T lymphocytes from wild type but not from NKG2D-deficient (Klrk1-/-) mice in vitro. The adoptive transfer of activated T lymphocytes from wild type donors induced a significantly reduced EAE disease in Klrk1-/- compared to wild type (Klrk1+/+) recipients. Characterization of T lymphocytes infiltrating the CNS of recipient mice shows that donor (CD45.1) rather than endogenous (CD45.2) CD4 T cells are the main producers of key cytokines (IFNγ, GM-CSF). In contrast, infiltrating CD8 T lymphocytes include mainly endogenous (CD45.2) cells exhibiting effector properties (NKG2D, granzyme B and IFNγ). Our data support the notion that endogenous CD8 T cells contribute to passive EAE pathobiology in a NKG2D-dependent manner. Collectively, our results point to the deleterious role of NKG2D and its MULT1 in the pathobiology of a MS mouse model.
-
Immunology and Microbiology
JunB is essential for IL-23-dependent pathogenicity of Th17 cells.
In Nature Communications on 30 May 2017 by Hasan, Z., Koizumi, S. I., et al.
CD4+ T-helper cells producing interleukin-17 (IL-17), known as T-helper 17 (TH17) cells, comprise heterogeneous subsets that exhibit distinct pathogenicity. Although pathogenic and non-pathogenic TH17 subsets share a common RORγt-dependent TH17 transcriptional programme, transcriptional regulatory mechanisms specific to each of these subsets are mostly unknown. Here we show that the AP-1 transcription factor JunB is critical for TH17 pathogenicity. JunB, which is induced by IL-6, is essential for expression of RORγt and IL-23 receptor by facilitating DNA binding of BATF at the Rorc locus in IL-23-dependent pathogenic TH17 cells, but not in TGF-β1-dependent non-pathogenic TH17 cells. Junb-deficient T cells fail to induce TH17-mediated autoimmune encephalomyelitis and colitis. However, JunB deficiency does not affect the abundance of gut-resident non-pathogenic TH17 cells. The selective requirement of JunB for IL-23-dependent TH17 pathogenicity suggests that the JunB-dependent pathway may be a therapeutic target for autoimmune diseases.
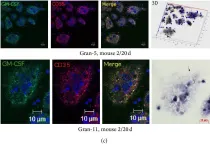
In J Immunol Res on 12 April 2016 by Ufimtseva, E.
Fig.10.C

-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from J Immunol Res by CiteAb, provided under a CC-BY license
Image 1 of 2
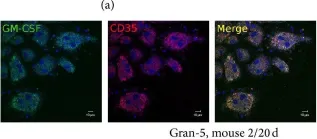
In J Immunol Res on 12 April 2016 by Ufimtseva, E.
Fig.10.A

-
ICC-IF
-
Mus musculus (House mouse)
Collected and cropped from J Immunol Res by CiteAb, provided under a CC-BY license
Image 1 of 2