
Alveolar macrophages (AMs) reside in the lower airways and play a crucial role in lung health and response to sterile inflammation and infections. AMs possess remarkable adaptability to different environmental challenges that can persist through their memory capacity (trained immunity). β-Glucan has been characterized as a potent inducer of central trained immunity by reprogramming haematopoietic stem cells in the bone marrow. In the present study, we show that systemic administration of β-glucan in mice induces peripheral trained immunity by reprogramming AMs in the lungs, in a Dectin1-independent manner. We furthermore demonstrate that AM reprogramming at both the transcriptional and metabolic levels exacerbate lung injury following bacterial (lipopolysaccharide) or viral (polyI:C) challenges via a neutrophil/IFN-γ-dependent manner. These findings identify an additional facet of β-glucan in trained immunity involving AM reprogramming and shed light on the potential detrimental effects of trained immunity.
© 2024, Prevel et al.
Product Citations: 311
β-Glucan reprograms alveolar macrophages via neutrophil/IFNγ axis in a murine model of lung injury.
In eLife on 8 July 2025 by Prevel, R., Pernet, E., et al.
In Nature Communications on 1 July 2025 by Perrotta, M., Perrotta, S., et al.
Activated immune cells infiltrate the vasculature during the pathophysiology of hypertension by establishing a vascular-immune interface that contributes to blood pressure dysregulation and organ failure. Many observations indicate a key role of CD8+ T cells in hypertension but mechanisms regulating their activation and interplay with the cardiovascular system are still unknown. In murine model, here we show that a specific member of the phosphoinositide-3-kinases (PI3K) family of lipid kinases, PI3Kγ, is a key intracellular signaling of CD8+ T cells activation and RANTES/CCL5 secretion in hypertension: CCL5-CCR5 signaling is crucial for the establishment of the vascular-immune interface in peripheral organs, lastly contributing to CD8+ tissue infiltration, organ dysfunction and blood pressure elevation. Our studies identify PI3Kγ as a booster of effector CD8+ T cell function, even in the absence of external stimuli. Lastly, an enhanced PI3Kγ signaling mediates the bystander activation of CD8+ T cells and proves effective in transferring the hypertensive phenotype between mice.
© 2025. The Author(s).
-
Cardiovascular biology
-
Immunology and Microbiology
In International Journal of Biological Sciences on 19 May 2025 by Bian, Z., Chen, J., et al.
Mutations in TP53, particularly the p.R248Q variant, contribute to the progression of castration-resistant prostate cancer (CRPC) by reshaping the tumor microenvironment (TME). This study examined the impact of p.R248Q (mutp53) on immune suppression and CRPC progression. We introduced the Trp53 p.R245Q mutation into RM-1 mouse prostate cancer (PCa) cells via CRISPR/Cas9, which mimics human TP53 p.R248Q. These cells were implanted into C57BL/6 mice to model tumor progression and immune interactions. Mice were treated with JAK2 and STAT3 inhibitors to assess immune and tumor responses. Tumor behavior and immune responses were analyzed via histology, immunofluorescence, flow cytometry, Enzyme-linked immunosorbent assay (ELISA), and bioinformatics. Findings were validated in the C4-2 human PCa cell line. Compared with wild-type p53, TP53 mutations were present in 27% of PCa patients and were significantly correlated with reduced overall survival (p < 0.001, HR = 1.97) and recurrence-free survival (p = 0.02, HR = 1.62). The p.R248Q mutation was most prevalent. Gene-edited mutp53 cells exhibited increased proliferation and tumorigenicity. Screening and validation confirmed that IL6/JAK2/STAT3 pathway activation in mutp53 tumors led to immune microenvironment alterations. Flow cytometry and immunofluorescence revealed an immunosuppressive profile, with decreased proinflammatory cytokines and elevated anti-inflammatory factors. Coimmunoprecipitation revealed that mutp53 competes with SHP1 for STAT3 binding, sustaining its activation. Inhibition of STAT3 reduced mutp53-driven immune suppression and tumor progression. Mutp53 promotes an immunosuppressive TME and facilitates CRPC progression through the STAT3 pathway, underscoring its potential as a therapeutic target.
© The author(s).
-
Cancer Research
PD-1 blockade employed at the time CD8+ T cells are activated enhances their antitumor efficacy.
In Journal for Immunotherapy of Cancer on 7 May 2025 by Moseman, J. E., Rastogi, I., et al.
We have previously shown that immune checkpoint receptors, including PD-1, are upregulated on T cells at the time of their activation, and that blockade of these receptors can improve the efficacy of antitumor vaccines. In the present study, we sought to determine whether, and by what mechanisms, the timing of PD-1 blockade with respect to vaccination affects antitumor T cell function.
TRAMP-C1 or E.G7-OVA tumor-bearing mice received PD-1 blockade at different timing intervals with a tumor-associated antigen vaccine. Tumor growth, survival, and immune-infiltrating populations were assessed. In vitro models of T cell activation using OT-I T cells and PD-(L)1 axis disruption with a PD-1 blocking antibody or PD-L1KO dendritic cells were used.
Mice receiving PD-1 blockade at the time of T cell activation with vaccine had better antitumor outcomes in comparison to mice receiving PD-1 blockade before or after immunization. T cells activated in vitro in the presence of PD-(L)1 axis disruption had a more differentiated, functional phenotype with decreased CD28 and CCR7 expression and increased production of the Tc1 cytokines IL-2, TNFα, and IFNγ. Intriguingly, a small subset of undifferentiated cells (CD28+) was of a stem-like Tc17 phenotype (IL-17α+, TCF1+). Tumor-bearing mice receiving T cells activated in the presence of PD-(L)1-axis disruption had better antitumor outcomes and a greater number of complete responses.
These data indicate that PD-1 blockade, when used with antitumor vaccines, acts primarily at the time of T cell activation, not exclusively within the tumor microenvironment. Consequently, PD-1 blockade may be best used when delivered concurrently with T cell activating agents such as vaccines.
© Author(s) (or their employer(s)) 2025. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ Group.
-
Immunology and Microbiology
Antitumor CD4+ T Helper 1 Cells Target and Control the Outgrowth of Disseminated Cancer Cells.
In Cancer Immunology Research on 2 May 2025 by Ganesan, R., Lee, M. C., et al.
Detection of disseminated cancer cells (DCC) in the bone marrow (BM) of patients with breast cancer is a critical predictor of late recurrence and distant metastasis. Conventional therapies often fail to completely eradicate DCCs in patients. In this study, we demonstrate that intratumoral priming of antitumor CD4+ T helper 1 (Th1) cells was able to eliminate the DCC burden in distant organs and prevent overt metastasis, independent of CD8+ T cells. Intratumoral priming of tumor antigen-specific CD4+ Th1 cells enhanced their migration to the BM and distant metastatic site to selectively target DCC burden. The majority of these intratumorally activated CD4+ T cells were CD4+PD1- T cells, supporting their nonexhaustion stage. Phenotypic characterization revealed enhanced infiltration of memory CD4+ T cells and effector CD4+ T cells in the primary tumor, tumor-draining lymph node, and DCC-driven metastasis site. A robust migration of CD4+CCR7+CXCR3+ Th1 cells and CD4+CCR7-CXCR3+ Th1 cells into distant organs further revealed their potential role in eradicating DCC-driven metastasis. The intratumoral priming of antitumor CD4+ Th1 cells failed to eradicate DCC-driven metastasis in CD4- or IFN-γ knockout mice. Moreover, antitumor CD4+ Th1 cells, by increasing IFN-γ production, inhibited various molecular aspects and increased classical and nonclassical MHC molecule expression in DCCs. This reduced stemness and self-renewal while increasing immune recognition in DCCs of patients with breast cancer. These results unveil an immune basis for antitumor CD4+ Th1 cells that modulate DCC tumorigenesis to prevent recurrence and metastasis in patients.
©2025 The Authors; Published by the American Association for Cancer Research.
-
Cancer Research
-
Immunology and Microbiology
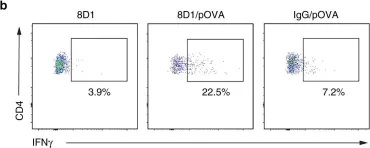
In Front Immunol on 23 July 2021 by Di Giorgio, E., Wang, L., et al.
Fig.5.E

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Front Immunol by CiteAb, provided under a CC-BY license
Image 1 of 3
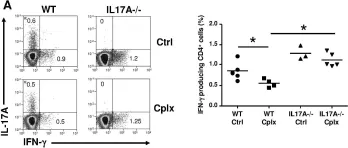
In Nat Commun on 21 February 2018 by Westhorpe, C. L. V., Norman, M. U., et al.
Fig.5.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 3
In PLoS One on 23 October 2013 by Vokaer, B., Charbonnier, L. M., et al.
Fig.5.A

-
FC/FACS
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 3