Tumour cells often evade immune pressure exerted by CD8+ T cells or immunotherapies through mechanisms that are largely unclear1,2. Here, using complementary in vivo and in vitro CRISPR-Cas9 genetic screens to target metabolic factors, we established voltage-dependent anion channel 2 (VDAC2) as an immune signal-dependent checkpoint that curtails interferon-γ (IFNγ)-mediated tumour destruction and inflammatory reprogramming of the tumour microenvironment. Targeting VDAC2 in tumour cells enabled IFNγ-induced cell death and cGAS-STING activation, and markedly improved anti-tumour effects and immunotherapeutic responses. Using a genome-scale genetic interaction screen, we identified BAK as the mediator of VDAC2-deficiency-induced effects. Mechanistically, IFNγ stimulation increased BIM, BID and BAK expression, with VDAC2 deficiency eliciting uncontrolled IFNγ-induced BAK activation and mitochondrial damage. Consequently, mitochondrial DNA was aberrantly released into the cytosol and triggered robust activation of cGAS-STING signalling and type I IFN response. Importantly, co-deletion of STING signalling components dampened the therapeutic effects of VDAC2 depletion in tumour cells, suggesting that targeting VDAC2 integrates CD8+ T cell- and IFNγ-mediated adaptive immunity with a tumour-intrinsic innate immune-like response. Together, our findings reveal VDAC2 as a dual-action target to overcome tumour immune evasion and establish the importance of coordinately destructing and inflaming tumours to enable efficacious cancer immunotherapy.
© 2025. The Author(s).
Product Citations: 55
VDAC2 loss elicits tumour destruction and inflammation for cancer therapy.
In Nature on 1 April 2025 by Yuan, S., Sun, R., et al.
-
Cancer Research
-
Immunology and Microbiology
In Vaccines on 3 March 2025 by Lv, Y., Tang, J., et al.
Background:Echinococcus granulosus represents a significant threat to animal husbandry and human health, but its consequences are often underestimated. Vaccination can prevent E. granulosus infection. We investigated the immune protective effect induced by the recombinant protein P29 of E. granulosus (rEg.P29) peptide vaccine. Methods: The CD4+ T-, CD8+ T-, Treg-, and CD8+CD107a+ T-cell proportions in the spleen and peripheral blood of infected mice were analyzed using flow cytometry. Additionally, we measured the proportions of IFN-γ and IL-2 secreted by memory T cells, CD19+CD138-B cells, CD19+CD138+ plasmablasts, CD19-CD138+ plasma cells, and CD19+IgD-IgG+ and CD19+IgD-IgA+ memory B cells. Results: No significant differences were noted in CD4+ T-, CD8+ T-, and CD8+CD107a+ Treg-cell percentages among the experimental groups. However, IFN-γ, IL-2, and TNF-α levels and vaccine-specific antibody concentrations in the plasma were significantly elevated in the rEg.P29T+B + CpG + infection and rEg.P29 + CpG + infection groups compared to those in the PBS + infection and CpG + infection groups. Similarly, CD19-CD138+ plasma cell and CD19+IgD-IgG+ and CD19+IgD-IgA+ memory B-cell populations, along with specific antibodies, were significantly higher in these groups. Especially, the average cyst burden in the rEg.P29T+B + CpG + infection and rEg.P29 + CpG + infection groups was significantly reduced compared to that in the PBS + infection and CpG + infection groups. Conclusions: Synthetic peptide vaccines targeting rEg.P29 can effectively inhibit cysts, offering a novel strategy for the development of vaccines against E. granulosus. These findings provide a foundation for further research on the immunogenicity and protective efficacy of rEg.P29-based vaccines.
-
Immunology and Microbiology
In IScience on 16 August 2024 by Fan, J., Zhang, Y., et al.
The immune evasion of emerging SARS-CoV-2 variants significantly undermines current vaccination efforts, calling for an updated vaccine composition. To identify optimal booster candidates against circulating JN.1, a panel of variant spikes were characterized. The omicron spikes exhibited reduced plasma membrane expression, accompanied by lower cell-cell fusion but increased viral entry. Regimens with DNA prime-DNA boost or DNA prime-adenoviral vectored vaccine boost by intramuscular immunization elicited neutralizing antibody (NAbs) and T cell responses against all variants except BA.2.86 and JN.1. Intranasal immunization induced high IgA and NAb titers in bronchoalveolar lavage against all variants except BA.2.86 and JN.1. T cell responses were generally comparable for all immunogens tested. JN.1 completely escaped NAbs in one immunized cohort, and breakthrough infections marginally boosted antibody titers. Overall, this study indicates intrinsic difficulty in eliciting NAbs against the JN.1 strain, whereas vaccines based on XBB and EG.5.1 are relatively superior in generating cross-reactive NAbs.
© 2024 The Authors.
-
Mus musculus (House mouse)
In Vaccines on 19 June 2024 by Han, R., Wang, T., et al.
Respiratory syncytial virus (RSV) is a leading cause of severe lower respiratory tract disease of infants and older people. There is an urgent need for safe and effective vaccines against RSV infection. In this study, we analyzed the effects of the immune response and protection with the RSV recombinant G protein extracellular domain (Gecto) combined with various adjuvants as novel subunit vaccines in mice. All groups receiving RSV Gecto combined with adjuvants exhibited robust humoral and cellular immunity compared to those receiving an adjuvant alone or inactivated RSV vaccine. The greatest effect was observed in mice receiving Gecto combined with a CpG ODN + Alum salt adjuvant, resulting in the highest production of neutralizing antibodies against both RSV A and B subtypes, G-specific IgG and IFN-γ production in splenocytes, and interleukin-2 and interferon-γ expression in CD4+ T cells. Significant humoral and cellular immune responses were observed in mice immunized with Gecto combined with AddaS03™ or cyclosporin A adjuvants. The vaccine containing the AddaS03™ adjuvant showed significantly high expression of interleukin-4 in CD4+ T cells. Cross-protection against a challenge with either RSV A or B subtypes was observed in the Gecto plus adjuvant groups, resulting in a significant decrease in viral load and reduced pathological damage in the mouse lungs. These findings offer valuable insights into the development and application of recombinant RSV G-subunit vaccines with adjuvants.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In Cell Reports on 26 December 2023 by Carreras-Sureda, A., Zhang, X., et al.
Store-operated Ca2+ entry (SOCE) mediated by stromal interacting molecule (STIM)-gated ORAI channels at endoplasmic reticulum (ER) and plasma membrane (PM) contact sites maintains adequate levels of Ca2+ within the ER lumen during Ca2+ signaling. Disruption of ER Ca2+ homeostasis activates the unfolded protein response (UPR) to restore proteostasis. Here, we report that the UPR transducer inositol-requiring enzyme 1 (IRE1) interacts with STIM1, promotes ER-PM contact sites, and enhances SOCE. IRE1 deficiency reduces T cell activation and human myoblast differentiation. In turn, STIM1 deficiency reduces IRE1 signaling after store depletion. Using a CaMPARI2-based Ca2+ genome-wide screen, we identify CAMKG2 and slc105a as SOCE enhancers during ER stress. Our findings unveil a direct crosstalk between SOCE and UPR via IRE1, acting as key regulator of ER Ca2+ and proteostasis in T cells and muscles. Under ER stress, this IRE1-STIM1 axis boosts SOCE to preserve immune cell functions, a pathway that could be targeted for cancer immunotherapy.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
-
Immunology and Microbiology
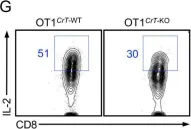
In J Exp Med on 2 December 2019 by Di Biase, S., Ma, X., et al.
Fig.2.G

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from J Exp Med by CiteAb, provided under a CC-BY license
Image 1 of 2
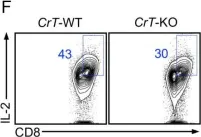
In J Exp Med on 2 December 2019 by Di Biase, S., Ma, X., et al.
Fig.3.F

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from J Exp Med by CiteAb, provided under a CC-BY license
Image 1 of 2