Acute myocardial infarction (MI) results in loss of cardiomyocytes and abnormal cardiac remodeling with severe inflammation and fibrosis. However, how cardiac repair can be achieved by timely resolution of inflammation and cardiac fibrosis remains incompletely understood. Our previous findings have shown that dual-specificity phosphatase 6 (DUSP6) is a regeneration repressor from zebrafish to rats. In this study, we found that intravenous administration of the DUSP6 inhibitor (E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one (BCI) improved heart function and reduced cardiac fibrosis in MI rats. Mechanistic analysis revealed that BCI attenuated macrophage inflammation through NF-κB and p38 signaling, independent of DUSP6 inhibition, leading to the downregulation of various cytokines and chemokines. In addition, BCI suppressed differentiation-related signaling pathways and decreased bone-marrow cell differentiation into macrophages through inhibiting DUSP6. Furthermore, intramyocardial injection of poly (D, L-lactic-co-glycolic acid)-loaded BCI after MI had a notable effect on cardiac repair. In summary, BCI improves heart function and reduces abnormal cardiac remodeling by inhibiting macrophage formation and inflammation post-MI, thus providing a promising pro-drug candidate for the treatment of MI and related heart diseases. This article has an associated First Person interview with the first author of the paper.© 2022. Published by The Company of Biologists Ltd.
Product Citations: 58
In Disease Models & Mechanisms on 1 May 2023 by Zhang, Z., Chen, Y., et al.
-
Mus musculus (House mouse)
-
Cardiovascular biology
-
Immunology and Microbiology
Dopamine receptor D2 confers colonization resistance via gut microbial metabolites
Preprint on BioRxiv : the Preprint Server for Biology on 14 March 2023 by Scott, S. A., Fu, J., et al.
Abstract/Summary Paragraph: The gut microbiome plays major roles in modulating host physiology. One such function is colonization resistance, or the ability of the microbial collective to protect the host against enteric pathogens 1–3 , including enterohemorrhagic Escherichia coli (EHEC) serotype O157:H7, an attaching and effacing (AE) food-borne pathogen that causes severe gastroenteritis, enterocolitis, bloody diarrhea, and acute renal failure (hemolytic uremic syndrome) 4,5 . Although gut microbes can provide colonization resistance by outcompeting some pathogens or modulating host defense provided by the gut barrier and intestinal immune cells, this phenomenon remains poorly understood. Emerging evidence suggests that small-molecule metabolites produced by the gut microbiota may mediate this process 6 . Here, we show that tryptophan (Trp)-derived metabolites produced by the gut bacteria protect the host against Citrobacter rodentium , a murine AE pathogen widely used as a model for EHEC infection 7,8 , by activation of the host neurotransmitter dopamine receptor D2 (DRD2) within the intestinal epithelium. We further find that these Trp metabolites act through DRD2 to decrease expression of a host actin regulatory protein involved in C. rodentium and EHEC attachment to the gut epithelium via formation of actin pedestals. Previously identified mechanisms of colonization resistance either directly affect the pathogen by competitive exclusion or indirectly by modulation of host defense mechanisms 9,10 , so our results delineate a noncanonical colonization resistance pathway against AE pathogens featuring an unconventional role for DRD2 outside the nervous system in controlling actin cytoskeletal organization within the gut epithelium. Our findings may inspire prophylactic and therapeutic approaches for improving gut health and treating gastrointestinal infections, which afflict millions globally.
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Cell Biology
In Cell Reports on 7 September 2021 by Lucas, R. M., Liu, L., et al.
Immune cells are armed with Toll-like receptors (TLRs) for sensing and responding to pathogens and other danger cues. The role of extracellular-signal-regulated kinases 1/2 (Erk1/2) in TLR signaling remains enigmatic, with both pro- and anti-inflammatory functions described. We reveal here that the immune-specific transmembrane adaptor SCIMP is a direct scaffold for Erk1/2 in TLR pathways, with high-resolution, live-cell imaging revealing that SCIMP guides the spatial and temporal recruitment of Erk2 to membrane ruffles and macropinosomes for pro-inflammatory TLR4 signaling. SCIMP-deficient mice display defects in Erk1/2 recruitment to TLR4, c-Fos activation, and pro-inflammatory cytokine production, with these effects being phenocopied by Erk1/2 signaling inhibition. Our findings thus delineate a selective role for SCIMP as a key scaffold for the membrane recruitment of Erk1/2 kinase to initiate TLR-mediated pro-inflammatory responses in macrophages.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In Cell Host & Microbe on 11 November 2020 by Liu, X., Boyer, M. A., et al.
Alveolar macrophages are among the first immune cells that respond to inhaled pathogens. However, numerous pathogens block macrophage-intrinsic immune responses, making it unclear how robust antimicrobial responses are generated. The intracellular bacterium Legionella pneumophila inhibits host translation, thereby impairing cytokine production by infected macrophages. Nevertheless, Legionella-infected macrophages induce an interleukin-1 (IL-1)-dependent inflammatory cytokine response by recruited monocytes and other cells that controls infection. How IL-1 directs these cells to produce inflammatory cytokines is unknown. Here, we show that collaboration with the alveolar epithelium is critical for controlling infection. IL-1 induces the alveolar epithelium to produce granulocyte-macrophage colony-stimulating factor (GM-CSF). Intriguingly, GM-CSF signaling amplifies inflammatory cytokine production in recruited monocytes by enhancing Toll-like receptor (TLR)-induced glycolysis. Our findings reveal that alveolar macrophages engage alveolar epithelial signals to metabolically reprogram monocytes for antibacterial inflammation.
Copyright © 2020 Elsevier Inc. All rights reserved.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In Immunology and Cell Biology on 1 August 2020 by Kapetanovic, R., Afroz, S. F., et al.
Mitochondria have a multitude of functions, including energy generation and cell signaling. Recent evidence suggests that mitochondrial dynamics (i.e. the balance between mitochondrial fission and fusion) also regulate immune functions. Here, we reveal that lipopolysaccharide (LPS) stimulation increases mitochondrial numbers in mouse bone marrow-derived macrophages (BMMs) and human monocyte-derived macrophages. In BMMs, this response requires Toll-like receptor 4 (Tlr4) and the TLR adaptor protein myeloid differentiation primary response 88 (MyD88) but is independent of mitochondrial biogenesis. Consistent with this phenomenon being a consequence of mitochondrial fission, the dynamin-related protein 1 (Drp1) GTPase that promotes mitochondrial fission is enriched on mitochondria in LPS-activated macrophages and is required for the LPS-mediated increase in mitochondrial numbers in both BMMs and mouse embryonic fibroblasts. Pharmacological agents that skew toward mitochondrial fusion also abrogated this response. LPS triggered acute Drp1 phosphorylation at serine 635 (S635), followed by sustained Drp1 dephosphorylation at serine 656 (S656), in BMMs. LPS-induced S656 dephosphorylation was abrogated in MyD88-deficient BMMs, suggesting that this post-translational modification is particularly important for Tlr4-inducible fission. Pharmacological or genetic targeting of Tlr4-inducible fission had selective effects on inflammatory mediator production, with LPS-inducible mitochondrial fission promoting the expression and/or secretion of a subset of inflammatory mediators in BMMs and mouse embryonic fibroblasts. Thus, triggering of Tlr4 results in MyD88-dependent activation of Drp1, leading to inducible mitochondrial fission and subsequent inflammatory responses in macrophages.
© 2020 The Authors. Immunology & Cell Biology published by John Wiley & Sons Australia, Ltd on behalf of Australian and New Zealand Society for Immunology, Inc.
-
ELISA
-
Mus musculus (House mouse)
-
Cell Biology
-
Immunology and Microbiology
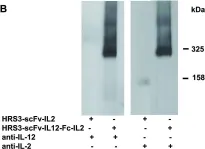
In PLoS One on 3 October 2012 by Jahn, T., Zuther, M., et al.
Fig.2.B

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 1