Cancer stem cells (CSCs), being the primary contributors in tumor initiation, metastasis, and relapse, ought to have seminal roles in evasion of immune surveillance. Tumor-promoting CD4+CD25+FOXP3+ T-regulatory cells (Tregs) have been described to abolish host defense mechanisms by impeding the activities of other immune cells including effector T cells. However, whether CSCs can convert effector T cells to immune-suppressive Treg subset, and if yes, the mechanism underlying CSC-induced Treg generation, are limitedly studied. In this regard, we observed a positive correlation between breast CSC and Treg signature markers in both in-silico and immunohistochemical analyses. Mirroring the conditions during tumor initiation, low number of CSCs could successfully generate CD4+CD25+FOXP3+ Treg cells from infiltrating CD4+ T lymphocytes in a contact-independent manner. Suppressing the proliferation potential as well as IFNγ production capacity of effector T cells, these Treg cells might be inhibiting antitumor immunity, thereby hindering immune-elimination of CSCs during tumor initiation. Furthermore, unlike non-stem cancer cells (NSCCs), CSCs escaped doxorubicin-induced apoptosis, thus constituting major surviving population after three rounds of chemotherapy. These drug-survived CSCs were also able to generate CD4+CD25+FOXP3+ Treg cells. Our search for the underlying mechanism further unveiled the role of CSC-shed immune-suppressive cytokine TGFβ, which was further increased by chemotherapy, in generating tumor Treg cells. In conclusion, during initiation as well as after chemotherapy, when NSCCs are not present in the tumor microenvironment, CSCs, albeit present in low numbers, generate immunosuppressive CD4+CD25+FOXP3+ Treg cells in a contact-independent manner by shedding high levels of immune-suppressive Treg-polarizing cytokine TGFβ, thus escaping immune-elimination and initiating the tumor or causing tumor relapse.
© 2023. The Author(s).
Product Citations: 22
In Discov Oncol on 1 December 2023 by Mukherjee, S., Chakraborty, S., et al.
-
Homo sapiens (Human)
-
Cancer Research
-
Immunology and Microbiology
-
Stem Cells and Developmental Biology
Protective role of cortistatin in pulmonary inflammation and fibrosis.
In British Journal of Pharmacology on 1 November 2021 by Barriga, M., Benitez, R., et al.
Acute lung injury (ALI), acute respiratory distress syndrome (ARDS) and pulmonary fibrosis remain major causes of morbidity, mortality and a healthcare burden in critically ill patient. There is an urgent need to identify factors causing susceptibility and for the design of new therapeutic agents. Here, we evaluate the effectiveness of the immunomodulatory neuropeptide cortistatin to regulate pulmonary inflammation and fibrosis in vivo.
ALI/ARDS and pulmonary fibrosis were induced experimentally in wild-type and cortistatin-deficient mice by pulmonary infusion of the bacterial endotoxin LPS or the chemotherapeutic drug bleomycin, and the histopathological signs, pulmonary leukocyte infiltration and cytokines, and fibrotic markers were evaluated.
Partially deficient mice in cortistatin showed exacerbated pulmonary damage, pulmonary inflammation, alveolar oedema and fibrosis, and subsequent increased respiratory failure and mortality when challenged to LPS or bleomycin, even at low doses. Treatment with cortistatin reversed these aggravated phenotypes and protected from progression to severe ARDS and fibrosis, after high exposure to both injury agents. Moreover, cortistatin-deficient pulmonary macrophages and fibroblasts showed exaggerated ex vivo inflammatory and fibrotic responses, and treatment with cortistatin impaired their activation. Finally, the protective effects of cortistatin in ALI and pulmonary fibrosis were partially inhibited by specific antagonists for somatostatin and ghrelin receptors.
We identified cortistatin as an endogenous inhibitor of pulmonary inflammation and fibrosis. Deficiency in cortistatin could be a marker of poor prognosis in inflammatory/fibrotic pulmonary disorders. Cortistatin-based therapies could emerge as attractive candidates to treat severe ALI/ARDS, including SARS-CoV-2-associated ARDS.
© 2021 The Authors. British Journal of Pharmacology published by John Wiley & Sons Ltd on behalf of British Pharmacological Society.
-
Cardiovascular biology
-
Immunology and Microbiology
-
Pharmacology
In Transplantation Direct on 1 October 2020 by Jankowska Gan, E., Agashe, V. V., et al.
Individuals harbor preexisting HLA-DR/DQ-restricted responses to collagen type V (ColV) mediated by Th17 cells under Treg control, both specific to peptides that bind to inherited HLA class II antigens. Yet after transplant, the donor-DR type somehow influences graft outcome. We hypothesized that, long after a lung or heart allograft, the particular HLA-DR type of the mismatched transplant donor transforms the specificity of the "anti-self" response. This could explain why, over long term, certain donor DRs could be more immunogenic than others.
We analyzed 7 HLA-DR15neg patients who had received a lung allograft from a DR15+ donor. To determine the mechanism of acquired specificity in self-reactivity, we analyzed the kinetics of DR1 (host) and DR15 (donor) peptide restriction in a heart transplant model using DR-transgenic mice.
Beyond 1.5 years post-lung transplant, all patients tested had acquired DR15-restricted immune responses to ColV peptides. These responses were either unrestrained Th17 type (n = 4) or Th17 controlled by Treg arising early (<5 y) or late (>7 y) after transplant (n = 4). Treg suppression via conventional (transforming growth factor-β [TGF-β]) and extracellular vesicle-associated (IL-35) cytokines correlated with superior outcomes. Naïve DR1 and DR15 transgenic mice had preexisting DR-restricted responses, exclusively to ColV fragments containing DR1- or DR15-binding peptides. When HLA-DR1 transgenic recipients of a HLA-DR15 heart developed ColV reactivity post-transplant, mice that acutely rejected (20-25 d) responded only to the DR1-restricted ColV peptide epitope. In animals whose grafts survived long term, we could detect acquisition of DR from the transplant donor onto the surface of recipient dendritic cells, and immune responses against a donor DR15-restricted ColV peptide.
These results might explain how certain donor HLA-DR types redirect host immune responses to novel peptides of critical self-antigens. Unless regulated, such responses may predispose the allograft to chronic rejection.
Copyright © 2020 The Author(s). Transplantation Direct. Published by Wolters Kluwer Health, Inc.
-
Cardiovascular biology
-
Immunology and Microbiology
In Cell Reports on 30 June 2020 by Yan, J., Pandey, S. P., et al.
IRF5 polymorphisms are associated with multiple immune-mediated diseases, including ulcerative colitis. IRF5 contributions are attributed to its role in myeloid lineages. How T cell-intrinsic IRF5 contributes to inflammatory outcomes is not well understood. We identify a previously undefined key role for T cell-intrinsic IRF5. In mice, IRF5 in CD4+ T cells promotes Th1- and Th17-associated cytokines and decreases Th2-associated cytokines. IRF5 is required for the optimal assembly of the TCR-initiated signaling complex and downstream signaling at early times, and at later times binds to promoters of Th1- and Th17-associated transcription factors and cytokines. IRF5 also regulates chemokine receptor-initiated signaling and, in turn, T cell migration. In vivo, IRF5 in CD4+ T cells enhances the severity of experimental colitis. Importantly, human CD4+ T cells from high IRF5-expressing disease-risk genetic carriers demonstrate increased chemokine-induced migration and Th1/Th17 cytokines and reduced Th2-associated and anti-inflammatory cytokines. These data demonstrate key roles for T cell-intrinsic IRF5 in inflammatory outcomes.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
-
ELISA
-
Mus musculus (House mouse)
-
Immunology and Microbiology
Regulatory Effect of Sishen Pill on Tfh Cells in Mice With Experimental Colitis.
In Frontiers in Physiology on 26 June 2020 by Liu, X. K., Zhao, H. M., et al.
The T follicular helper T (Tfh) cells play a significant role in the pathogenesis of inflammatory bowel disease (IBD), which is regulated by the Bcl-6/Blimp-1 pathway. Some studies have suggested that regulating activation of the Bcl-6/Blimp-1 pathway should be an effective method to treat IBD. Sishen Pill (SSP) has been used frequently to treat chronic colitis. Its mechanism is related to the downstream proteins in the Bcl-6/Blimp-1 pathway. However, it is unknown whether SSP regulates the function and differentiation of Tfh cells to treat IBD. In the present study, chronic colitis was induced by dextran sodium sulfate and treated with SSP for 7 days. SSP effectively treated chronic colitis, regulated the balance between Tfh10, Tfh17 and T follicular regulatory cells, while SSP increased the Blimp-1 level, inhibited expressions of Bcl-6, T-cell costimulator, programmed death (PD)-1 and PD-ligand 1 on the surface of Tfh cells. SSP inhibited activation of BcL-6, phosphorylated signal transducer and activator of transcription (p-STAT)3, signal lymphocyte activation molecule (SLAM)-associated protein but improved Blimp-1 and STAT3 expression in colonic tissues. The results indicated that SSP regulated the differentiation and function of Tfh cells to treat IBD, which was potentially related with inhibiting the Bcl-6/Blimp-1 pathway.
Copyright © 2020 Liu, Zhao, Wang, Ge, Zhong, Long and Liu.
-
ELISA
-
Mus musculus (House mouse)
-
Endocrinology and Physiology
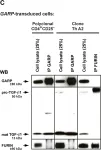
In PLoS One on 8 October 2013 by Gauthy, E., Cuende, J., et al.
Fig.3.C

-
WB post IP
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 1