The von Hippel-Lindau (VHL) tumor suppressor is a substrate-defining component of E3 ubiquitin ligase complexes that target cellular substrates for proteasome-mediated degradation. VHL inactivation by mutation or transcriptional silencing is observed in most sporadic cases of clear cell renal cell carcinoma (ccRCC). VHL loss in ccRCC leads to constitutive stabilization of E3 ligase substrates, including hypoxia inducible factor α (HIFα). HIFα stabilization upon VHL loss is known to contribute to ccRCC development through transactivation of hypoxia-responsive genes. HIF-independent VHL targets have been implicated in oncogenesis, although those mechanisms are less well-defined than for HIFα. Using proximity labeling to identify proteasomal-sensitive VHL interactors, we identified retinoblastoma protein (pRb) as a novel substrate of VHL. Mechanistically, VHL interacts with pRb in a proteasomal-sensitive manner, promoting its ubiquitin-mediated degradation. Concordantly, VHL-inactivation results in pRb hyperstabilization. Functionally, loss of pRb in ccRCC led to increased cell death, transcriptional changes, and loss of oncogenic properties in vitro and in vivo. We also show that downstream transcriptional changes induced by pRb hyperstabilization may contribute to ccRCC tumor development. Together, our findings reveal a novel VHL-related pathway which can be therapeutically targeted to inhibit ccRCC tumor development.
© 2025. The Author(s).
Product Citations: 52
Loss of VHL-mediated pRb regulation promotes clear cell renal cell carcinoma.
In Cell Death & Disease on 16 April 2025 by Akuma, M., Kim, M., et al.
-
Cancer Research
-
Cell Biology
In Nature Metabolism on 1 June 2022 by Li, S., Li, W., et al.
Mitochondria are the main consumers of oxygen within the cell. How mitochondria sense oxygen levels remains unknown. Here we show an oxygen-sensitive regulation of TFAM, an activator of mitochondrial transcription and replication, whose alteration is linked to tumours arising in the von Hippel-Lindau syndrome. TFAM is hydroxylated by EGLN3 and subsequently bound by the von Hippel-Lindau tumour-suppressor protein, which stabilizes TFAM by preventing mitochondrial proteolysis. Cells lacking wild-type VHL or in which EGLN3 is inactivated have reduced mitochondrial mass. Tumorigenic VHL variants leading to different clinical manifestations fail to bind hydroxylated TFAM. In contrast, cells harbouring the Chuvash polycythaemia VHLR200W mutation, involved in hypoxia-sensing disorders without tumour development, are capable of binding hydroxylated TFAM. Accordingly, VHL-related tumours, such as pheochromocytoma and renal cell carcinoma cells, display low mitochondrial content, suggesting that impaired mitochondrial biogenesis is linked to VHL tumorigenesis. Finally, inhibiting proteolysis by targeting LONP1 increases mitochondrial content in VHL-deficient cells and sensitizes therapy-resistant tumours to sorafenib treatment. Our results offer pharmacological avenues to sensitize therapy-resistant VHL tumours by focusing on the mitochondria.
© 2022. The Author(s).
-
WB
-
Cell Biology
In Frontiers in Endocrinology on 8 April 2022 by Mathó, C., Fernandez, M. C., et al.
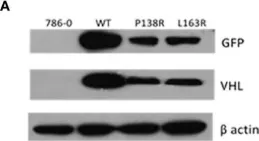
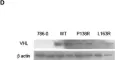
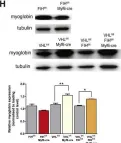
The von Hippel-Lindau (VHL) disease is an autosomal dominant cancer syndrome caused by mutations in the VHL tumor suppressor gene. VHL protein (pVHL) forms a complex (VBC) with Elongins B-C, Cullin2, and Rbx1. Although other functions have been discovered, the most described function of pVHL is to recognize and target hypoxia-inducible factor (HIF) for degradation. This work comprises the functional characterization of two novel variants of the VHL gene (P138R and L163R) that have been described in our center in patients with VHL disease by in vitro, in vivo, and in silico approaches. In vitro, we found that these variants have a significantly shorter half-life compared to wild-type VHL but still form a functional VBC complex. Altered fibronectin deposition was evidenced for both variants using immunofluorescence. In vivo studies revealed that both variants failed to suppress tumor growth. By means of molecular dynamics simulations, we inspected in silico the nature of the changes introduced by each variant in the VBC complex. We have demonstrated the pathogenicity of P138R and L163R novel variants, involving HIF-dependent and HIF-independent mechanisms. These results provide the basis for future studies regarding the impact of structural alterations on posttranslational modifications that drive pVHL's fate and functions.
Copyright © 2022 Mathó, Fernández, Bonanata, Liu, Martin, Vieites, Sansó, Barontini, Jonasch, Coitiño and Pennisi.
-
WB
-
Endocrinology and Physiology
ER-stress promotes VHL-independent degradation of hypoxia-inducible factors via FBXW1A/βTrCP.
In Redox Biology on 1 April 2022 by Mennerich, D., Kubaichuk, K., et al.
Metabolic adaptation and signal integration in response to hypoxic conditions is mainly regulated by hypoxia-inducible factors (HIFs). At the same time, hypoxia induces ROS formation and activates the unfolded protein response (UPR), indicative of endoplasmic reticulum (ER) stress. However, whether ER stress would affect the hypoxia response remains ill-defined. Here we report that feeding mice a high fat diet causes ER stress and attenuates the response to hypoxia. Mechanistically, ER stress promotes HIF-1α and HIF-2α degradation independent of ROS, Ca2+, and the von Hippel-Lindau (VHL) pathway, involving GSK3β and the ubiquitin ligase FBXW1A/βTrCP. Thereby, we reveal a previously unknown function of the GSK3β/HIFα/βTrCP1 axis in ER homeostasis and demonstrate that inhibition of the HIF-1 and HIF-2 response and genetic deficiency of GSK3β affects proliferation, migration, and sensitizes cells for ER stress promoted apoptosis. Vice versa, we show that hypoxia affects the ER stress response mainly through the PERK-arm of the UPR. Overall, we discovered previously unrecognized links between the HIF pathway and the ER stress response and uncovered an essential survival pathway for cells under ER stress.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
In Cancers on 2 August 2021 by Buart, S., Terry, S., et al.
Von Hippel-Lindau disease (VHL) is a rare hereditary syndrome due to mutations of the VHL tumor suppressor gene. Patients harboring the R167Q mutation of the VHL gene have a high risk of developing ccRCCs. We asked whether the R167Q mutation with critical aspects of pseudo-hypoxia interferes with tumor plasticity. For this purpose, we used wild-type VHL (WT-VHL) and VHL-R167Q reconstituted cells. We showed that WT-VHL and VHL-R167Q expression had a similar effect on cell morphology and colony formation. However, cells transfected with VHL-R167Q display an intermediate, HIF2-dependent, epithelial-mesenchymal phenotype. Using RNA sequencing, we showed that this mutation upregulates the expression of genes involved in the hypoxia pathway, indicating that such mutation is conferring an enhanced pseudo-hypoxic state. Importantly, this hypoxic state correlates with the induction of genes belonging to epithelial-mesenchymal transition (EMT) and stemness pathways, as revealed by GSEA TCGA analysis. Moreover, among these deregulated genes, we identified nine genes specifically associated with a poor patient survival in the TCGA KIRC dataset. Together, these observations support the hypothesis that a discrete VHL point mutation interferes with tumor plasticity and may impact cell behavior by exacerbating phenotypic switching. A better understanding of the role of this mutation might guide the search for more effective treatments to combat ccRCCs.
-
WB
-
IHC
-
Cancer Research
In Front Endocrinol (Lausanne) on 8 April 2022 by Mathó, C., Fernandez, M. C., et al.
Fig.1.A

-
WB
-
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 5
In Front Endocrinol (Lausanne) on 8 April 2022 by Mathó, C., Fernandez, M. C., et al.
Fig.4.D

-
WB
-
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 5
In Front Oncol on 20 October 2018 by Briston, T., Stephen, J. M., et al.
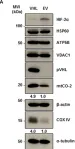
Fig.1.A

-
WB
-
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 5
In Front Oncol on 20 October 2018 by Briston, T., Stephen, J. M., et al.
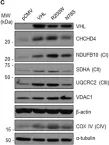
Fig.4.C

-
WB
-
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 5
In Cell Metab on 3 April 2018 by Sim, J., Cowburn, A. S., et al.
Fig.5.H

-
WB
-
Collected and cropped from Cell Metab by CiteAb, provided under a CC-BY license
Image 1 of 5