Skeletal muscle regeneration occurs through the finely timed activation of resident muscle stem cells (MuSC). Following injury, MuSC exit quiescence, undergo myogenic commitment, and regenerate the muscle. This process is coordinated by tissue microenvironment cues, however the underlying mechanisms regulating MuSC function are still poorly understood. Here, we demonstrate that the extracellular matrix protein Tenascin-C (TnC) promotes MuSC self-renewal and function. Mice lacking TnC exhibit reduced number of MuSC, and defects in MuSC self-renewal, myogenic commitment, and repair. We show that fibro-adipogenic progenitors are the primary cellular source of TnC during regeneration, and that MuSC respond through the surface receptor Annexin A2. We further demonstrate that TnC declines during aging, leading to impaired MuSC function. Aged MuSC exposed to soluble TnC show a rescued ability to both migrate and self-renew in vitro. Overall, our results highlight the pivotal role of TnC during muscle repair in healthy and aging muscle.
© 2025. The Author(s).
Product Citations: 90
In Communications Biology on 5 December 2025 by Cecchini, A., Loreti, M., et al.
-
Stem Cells and Developmental Biology
Impaired myogenesis in limb girdle muscular dystrophy type 2B.
In Scientific Reports on 30 September 2025 by Souza, L. S., Ishiba, R., et al.
The skeletal muscle tissue has a remarkable capacity of growth and regeneration. Fusion of myoblasts and myotubes elongation are fundamental processes in muscle development. Previous studies have depicted impaired myogenic processes in animal models and myoblast from human patients with muscle diseases. Here, we evaluated the myogenesis in patients with Limb-girdle Muscle Dystrophy 2B (LGMD2B). Aiming to explain why dysferlin-deficient muscle cells lose its myogenic potential, we used immortalized myoblasts from LGMD2B patients and a cellular DYSF knocking-out model. Myotubes from patients were smaller and containing less myonuclei than control myotubes. Main muscle regulatory factors expression were not altered in these cells. The analysis of the expression of newly described genes associated with muscle fusion and growth, such as MYMK, MYMX, PALMD, SHISA2, COL25A, didn´t show any difference with controls, which is a novel finding. It was also observed that dysferlin deficiency doesn't alter the expression of FAM65B and HDAC6 genes, components of a proposed protein complex that needs to be formed to allow muscle differentiation. Interestingly, morphometric analysis of DYSF knock-out myotubes induced by CRISPR/Cas9 also revealed reduced myogenic capacity with formation of smaller myotubes. These findings suggest that the absence of DYSF itself is sufficient to impair muscle formation in vitro, and that downstream gene and protein expression related to muscle development might depend on the presence and proper function of dysferlin.
© 2025. The Author(s).
Effect of lactation on postpartum pelvic floor muscle regeneration in preclinical model.
In Npj Women's Health on 16 June 2025 by Boscolo Sesillo, F., Manoochehri, H., et al.
Pelvic floor muscle (PFM) recovery following childbirth is essential for preserving pelvic floor function. Despite this, the impact of parturition and lactation on pelvic muscle stem cells (MuSCs), indispensable for skeletal muscle maintenance and regeneration, remains unknown. We determined that vaginal delivery does not cause mechanical injury of the rat PFMs, enabling us to uncouple the effects of lactation on muscle homeostasis from PFM regeneration following simulated birth injury (SBI). Tibialis anterior (TA) served as non-pelvic control. This novel study demonstrates that in the absence of birth injury, lactation blocks MuSC proliferation in PFM and TA, suggesting that postpartum systemic milieu affects MuSCs in pelvic and non-pelvic muscles. In contrast, SBI negated the inhibitory effect of lactation on MuSCs in PFM but not in TA, indicating that local signals released by the injured muscle overcome systemic inhibitory effects of lactation, which persist in muscles remote from the site of injury.
© The Author(s) 2025.
Epigenetic control of myogenic identity of human muscle stem cells in Duchenne muscular dystrophy.
In IScience on 20 December 2024 by Massenet, J., Weiss-Gayet, M., et al.
In Duchenne muscular dystrophy (DMD), muscle stem cells' (MuSCs) regenerative capacities are overwhelmed leading to fibrosis. Whether MuSCs have intrinsic defects or are disrupted by their environment is unclear. We investigated cell behavior and gene expression of MuSCs from DMD or healthy human muscles. Proliferation, differentiation, and fusion were unaltered in DMD-MuSCs, but with time, they lost their myogenic identity twice as fast as healthy MuSCs. The rapid drift toward a fibroblast-like cell identity was observed at the clonal level, and resulted from altered expression of epigenetic enzymes. Re-expression of CBX3, SMC3, H2AFV, and H3F3B prevented the MuSC identity drift. Among epigenetic changes, a closing of chromatin at the transcription factor MEF2B locus caused downregulation of its expression and loss of the myogenic fate. Re-expression of MEF2B in DMD-MuSCs restored their myogenic fate. MEF2B is key in the maintenance of myogenic identity in human MuSCs, which is altered in DMD.
© 2024 The Authors.
-
Homo sapiens (Human)
-
Genetics
-
Stem Cells and Developmental Biology
StarD7 deficiency switches on glycolysis and promotes mitophagy flux in C2C12 myoblasts.
In The FEBS Journal on 1 January 2024 by Rojas, M. L., Muñoz, J. P., et al.
StarD7 is a member of the START protein family required for phosphatidylcholine delivery to the mitochondria, thus key to maintain mitochondrial structure. Its deficiency has been associated with an impairment of cellular processes, such as proliferation and migration, and it has also been reported that it is needed in myogenic differentiation. Here, we show that StarD7 deficiency in C2C12 muscle cells results in the accumulation of abnormal mitochondria, a reduced number of mitochondria per cell area and increased glycolysis. In addition, StarD7-deficient cells undergo an increase in mitochondria-ER contact sites, reduced connexin 43 expression, and disturbances in lipid handling, evidenced by lipid droplet accumulation and decreased levels in phosphatidylserine synthase 1 and 2 expression. Interestingly, StarD7-deficient cells showed alterations in mitophagy markers. We observed accumulation of LC3B-II and BNIP3 proteins in mitochondria-enriched fractions and accumulation of autophagolysosomal and lysosomal vesicles in StarD7-deficient cells. Furthermore, live-cell imaging experiments of StarD7 knockdown cells expressing mitochondria-targeted mKeima indicated an enhanced mitochondria delivery into lysosomes. Importantly, StarD7 reconstitution in StarD7-deficient cells restores LC3B-II expression in mitochondria-enriched fractions at similar levels to those observed in control cells. Collectively, these findings suggest that StarD7-deficient C2C12 myoblasts are associated with altered cristae structure, disturbances in neutral lipid accumulation, glucose metabolism, and increased mitophagy flux. The alterations mentioned above allow for the maintenance of mitochondrial function.
© 2023 Federation of European Biochemical Societies.
-
WB
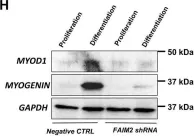
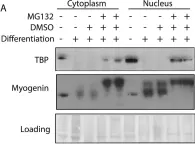
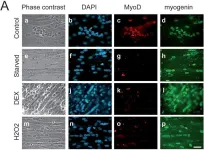
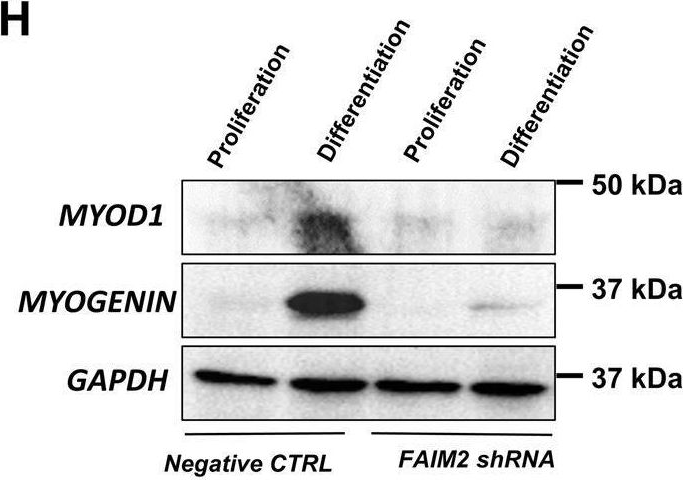
In Cell Death Dis on 25 April 2022 by Soliman, H. A. N., Toso, E. A., et al.
Fig.5.H
-
WB
-
Collected and cropped from Cell Death & Disease by CiteAb, provided under a CC-BY license
Image 1 of 17
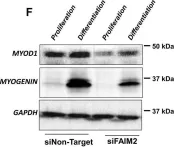
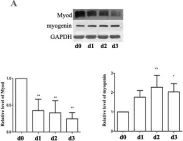
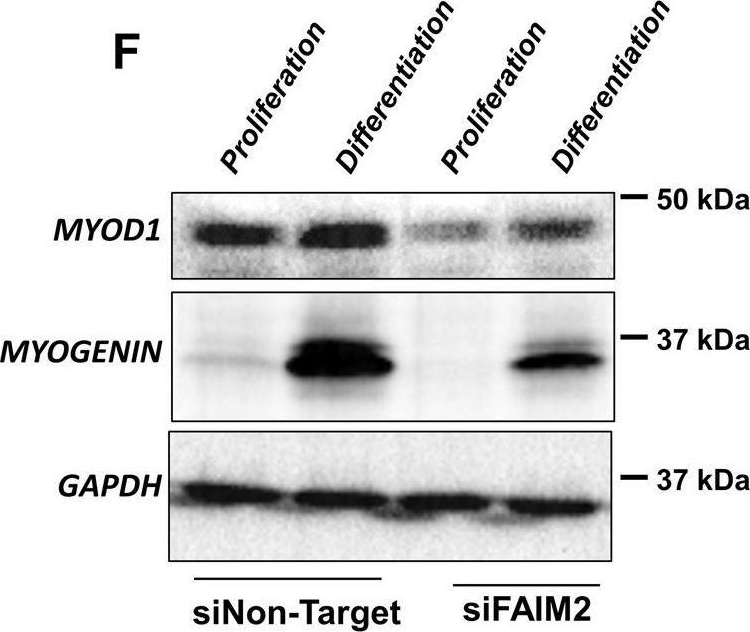
In Cell Death Dis on 25 April 2022 by Soliman, H. A. N., Toso, E. A., et al.
Fig.4.F
-
WB
-
Collected and cropped from Cell Death & Disease by CiteAb, provided under a CC-BY license
Image 1 of 17
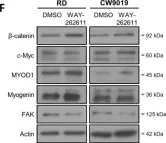
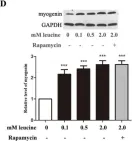
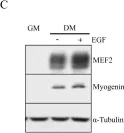
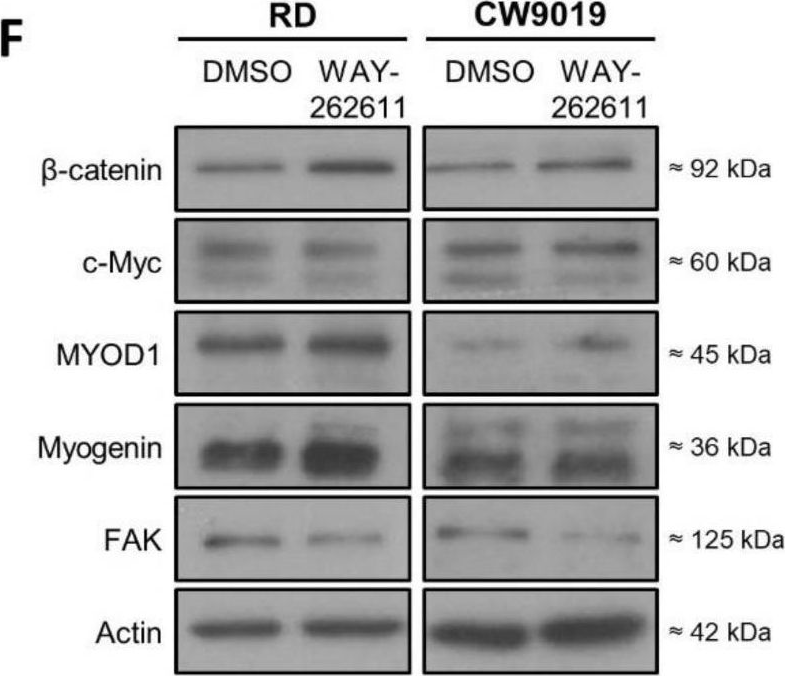
In Int J Mol Sci on 29 November 2021 by Giralt, I., Gallo-Oller, G., et al.
Fig.3.F
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 17
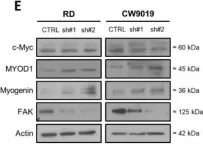
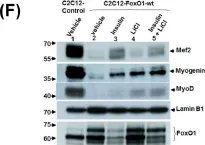
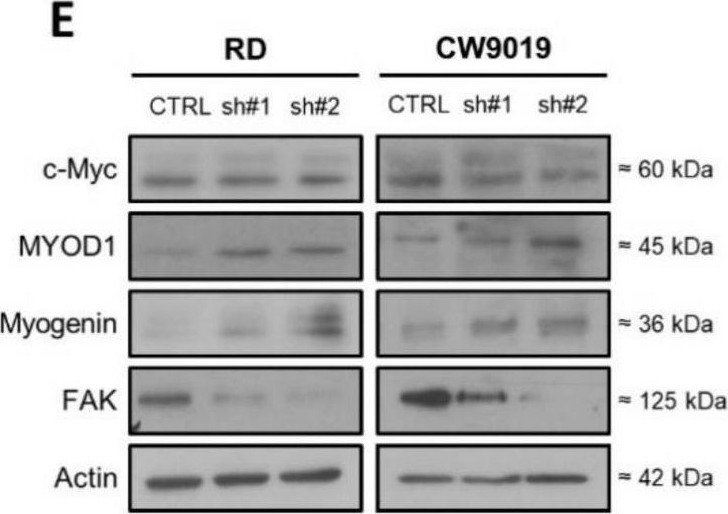
In Int J Mol Sci on 29 November 2021 by Giralt, I., Gallo-Oller, G., et al.
Fig.2.E
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 17
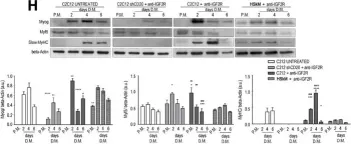
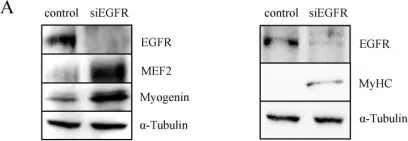
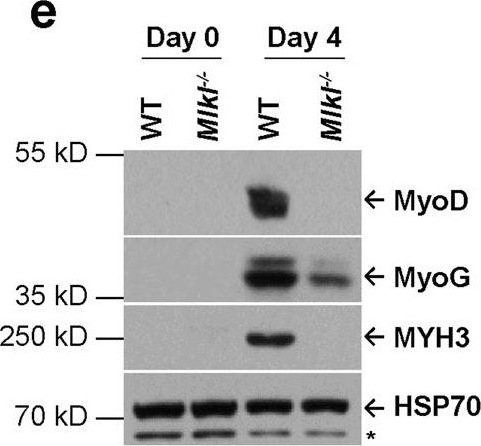
In Cell Res on 1 December 2020 by Zhou, S., Zhang, W., et al.
Fig.1.E
-
WB
-
Collected and cropped from Cell Research by CiteAb, provided under a CC-BY license
Image 1 of 17
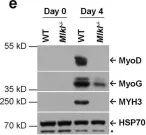
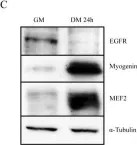
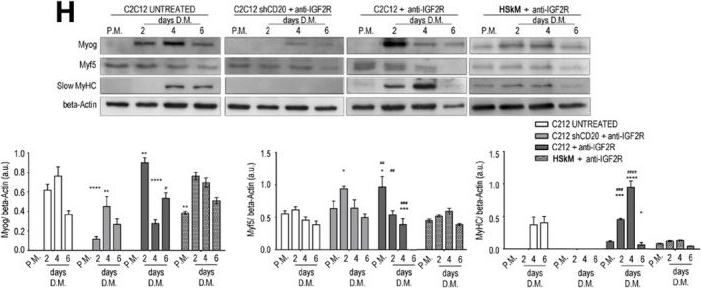
In EMBO Mol Med on 9 January 2020 by Bella, P., Farini, A., et al.
Fig.2.H
-
WB
-
Mus musculus (House mouse)
Collected and cropped from EMBO Molecular Medicine by CiteAb, provided under a CC-BY license
Image 1 of 17
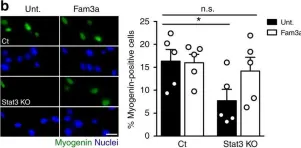
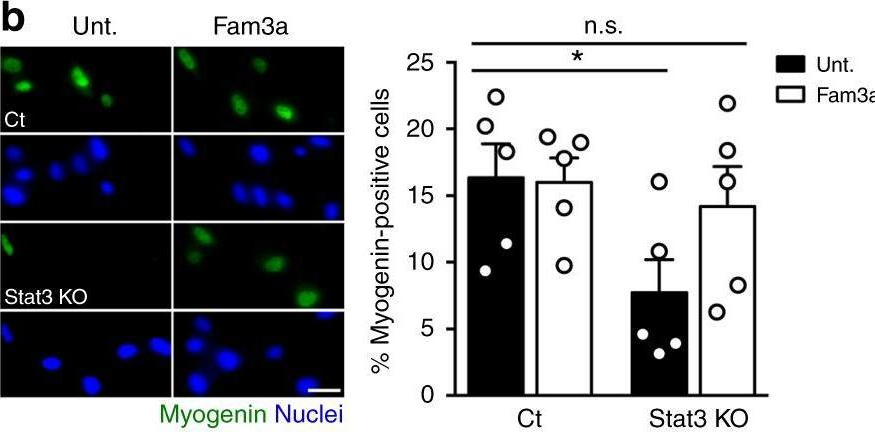
In Nat Commun on 17 April 2019 by Sala, D., Cunningham, T. J., et al.
Fig.6.B
-
ICC-IF
-
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 17
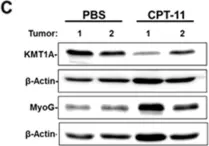
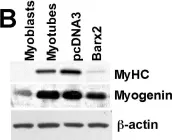
In Oncotarget on 25 May 2018 by Wolff, D. W., Lee, M. H., et al.
Fig.3.C
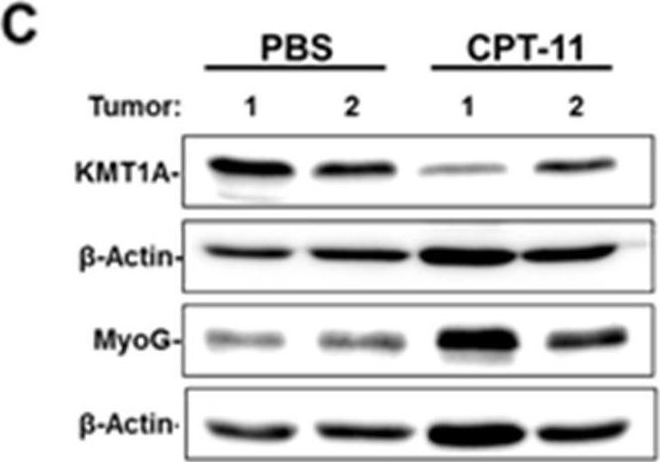
-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 17
In Elife on 22 September 2015 by Li, L., Martinez, S. S., et al.
Fig.1.A
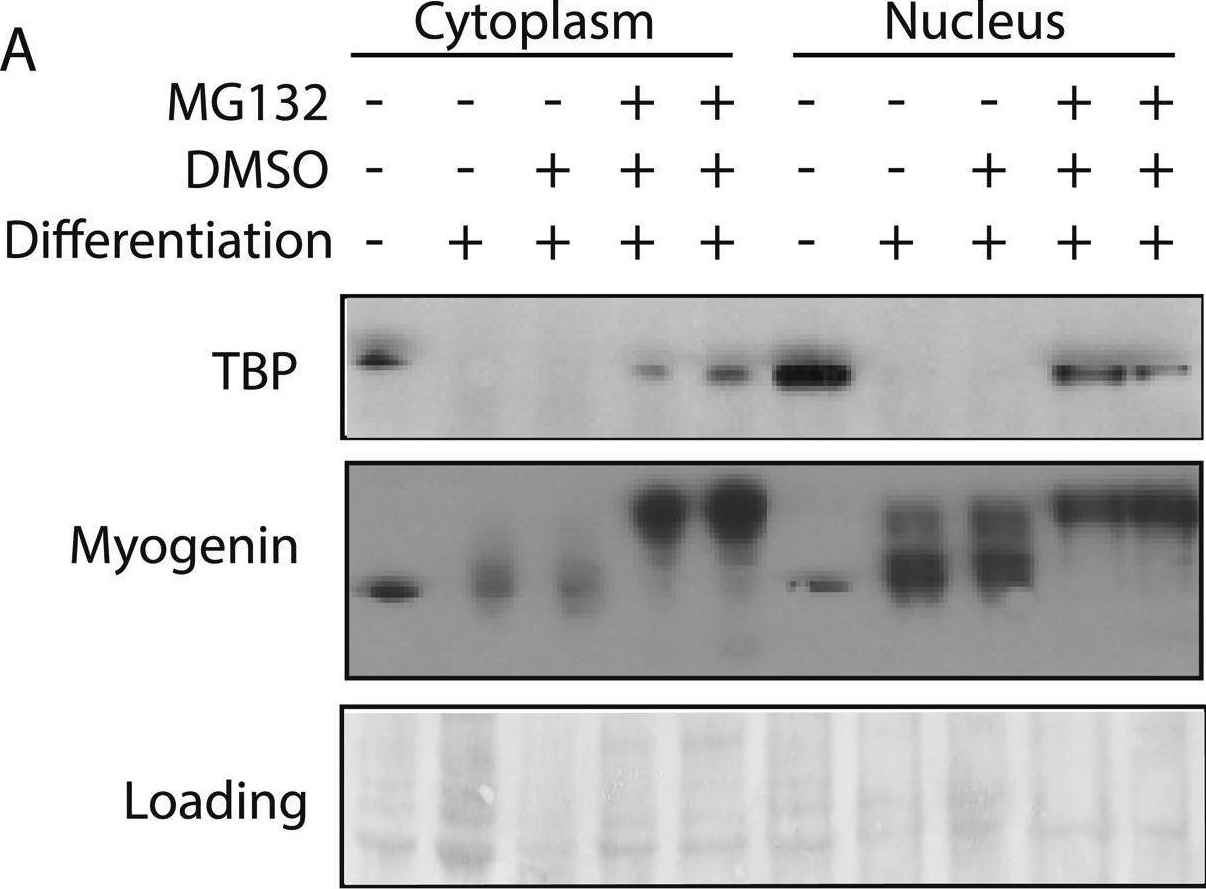
-
WB
-
Collected and cropped from eLife by CiteAb, provided under a CC-BY license
Image 1 of 17
In Nutrients on 8 May 2015 by Dai, J. M., Yu, M. X., et al.
Fig.5.A
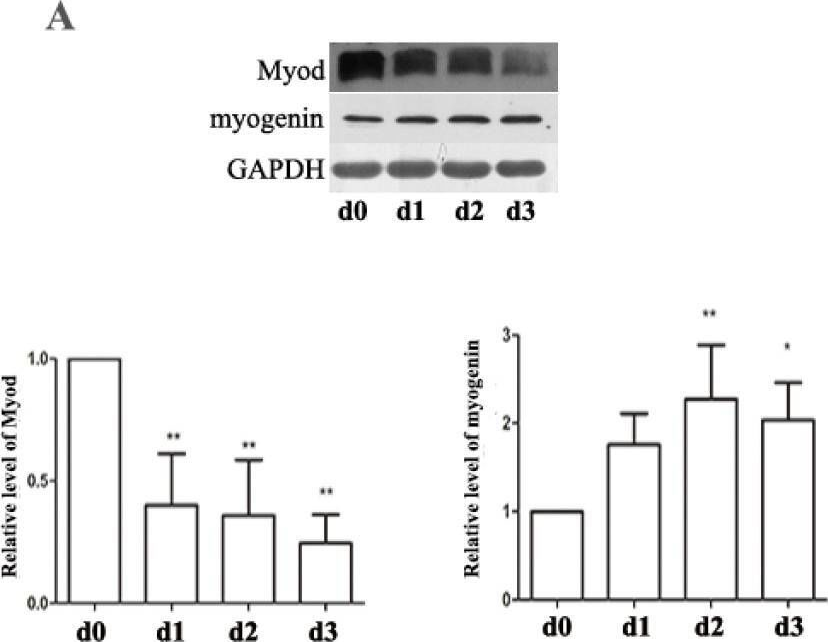
-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Nutrients by CiteAb, provided under a CC-BY license
Image 1 of 17
In Nutrients on 8 May 2015 by Dai, J. M., Yu, M. X., et al.
Fig.5.D
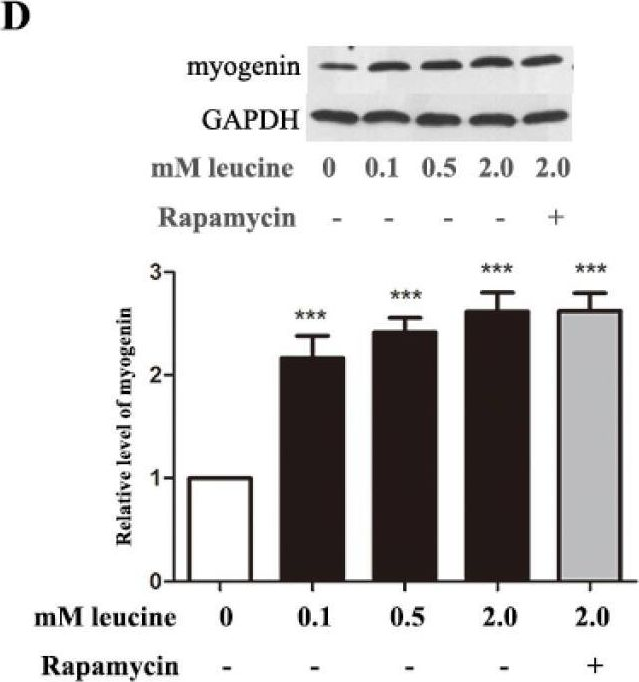
-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Nutrients by CiteAb, provided under a CC-BY license
Image 1 of 17
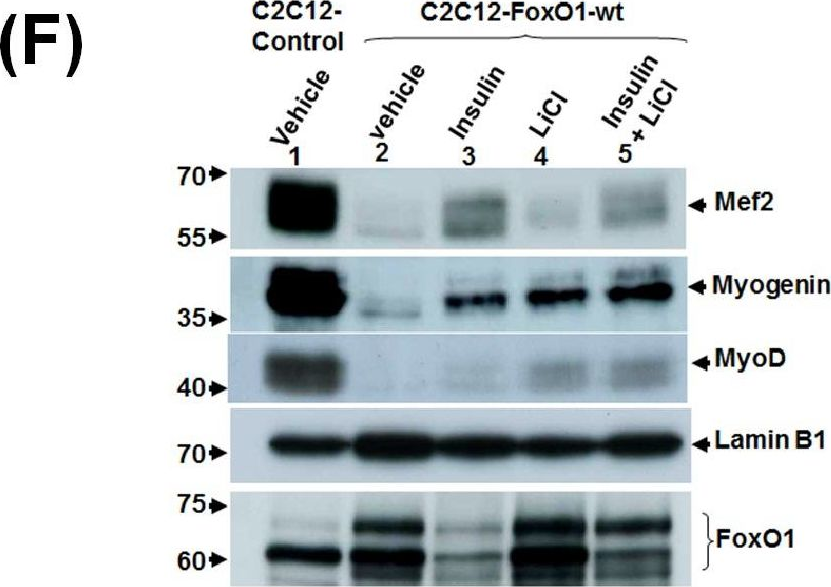
In PLoS One on 20 February 2014 by Wu, Y. J., Fang, Y. H., et al.
Fig.3.F
-
WB
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17
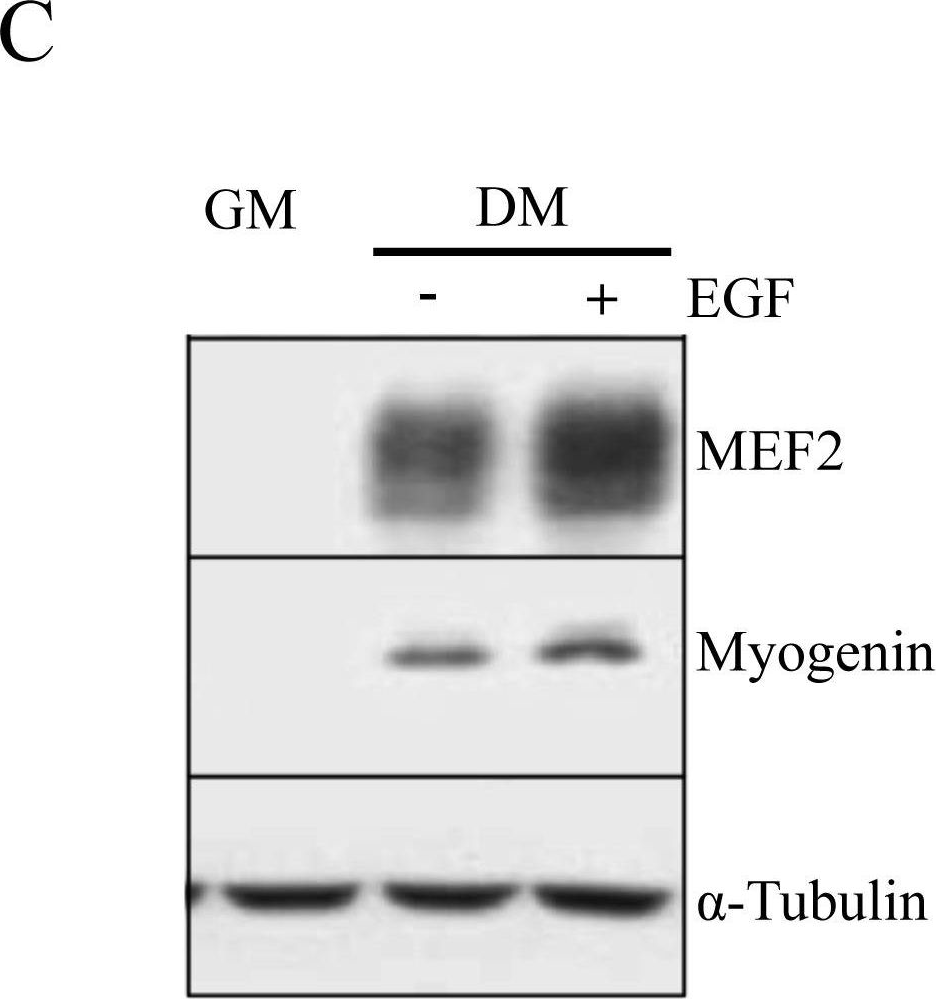
In PLoS One on 24 August 2013 by Leroy, M. C., Perroud, J., et al.
Fig.6.C
-
WB
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17
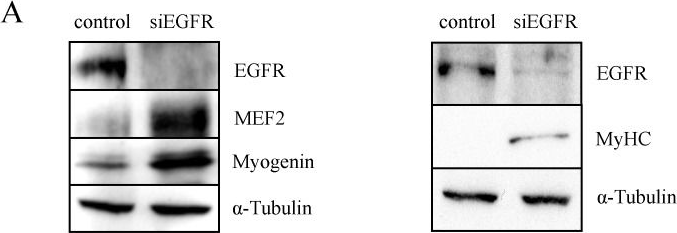
In PLoS One on 24 August 2013 by Leroy, M. C., Perroud, J., et al.
Fig.3.A
-
WB
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17
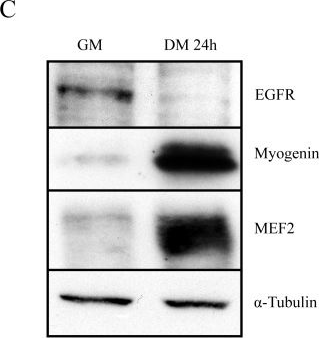
In PLoS One on 24 August 2013 by Leroy, M. C., Perroud, J., et al.
Fig.1.C
-
WB
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17
In PLoS One on 27 July 2010 by Meech, R., Gomez, M., et al.
Fig.1.B
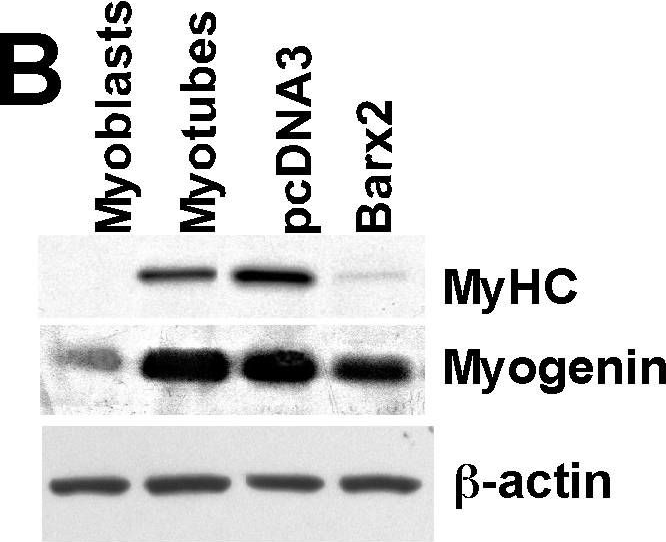
-
WB
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17
In PLoS One on 26 March 2009 by Lagirand-Cantaloube, J., Cornille, K., et al.
Fig.3.A
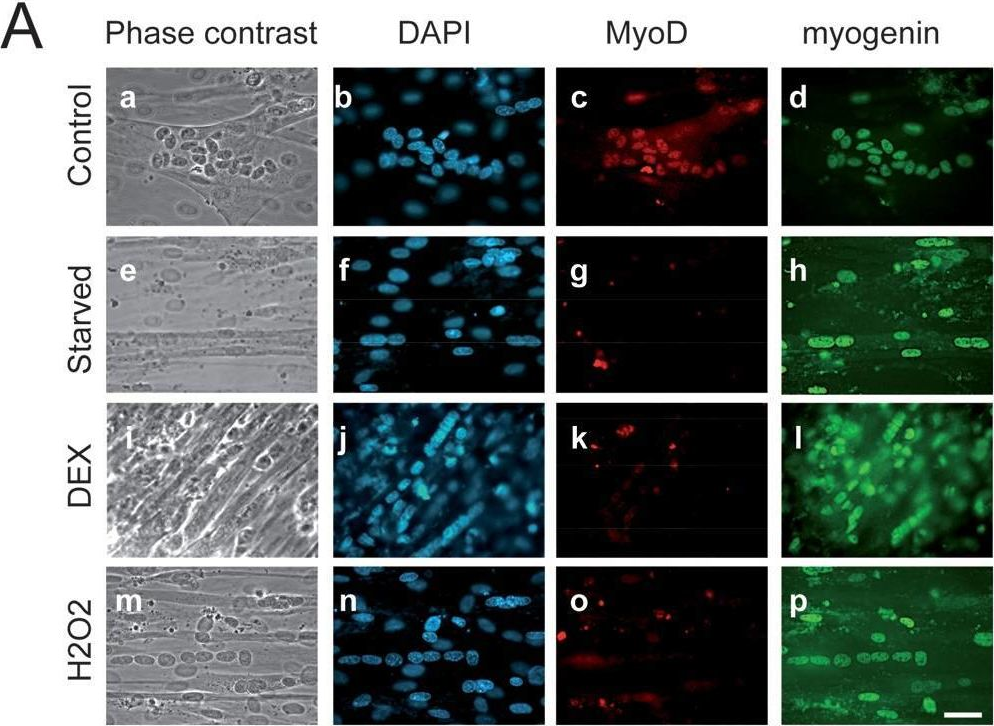
-
ICC-IF
-
Collected and cropped from PLoS ONE by CiteAb, provided under a CC-BY license
Image 1 of 17