
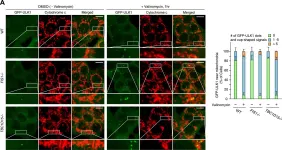
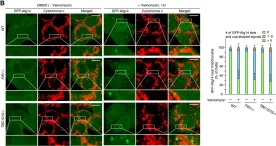
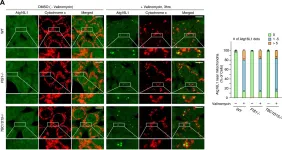
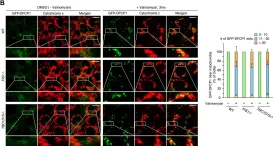
Neuronal death in acute ischemic stroke (AIS) is largely caused by the neurotoxic mechanism of oxidative/nitrosative stress, which is responsible for ischemia-reperfusion injury. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a multifunctional protein that mediates cell death under oxidative/nitrosative stress. The active site of GAPDH, residue cysteine-152 (Cys-152), is oxidized and forms intermolecular disulfide bonds that induce GAPDH aggregation, which causes mitochondrial dysfunction and eventually leads to cell death. A GAPDH-C152A mutant dominant-negatively suppresses GAPDH aggregation. Herein, we report that neuron-specific expression of GAPDH-C152A in conditional transgenic mice decreased GAPDH aggregation and brain damage induced by ischemia-reperfusion injury in an AIS mouse model. Furthermore, GAPDH aggregation inhibitor peptide-17, developed by our peptide-screening-methods, ameliorated brain infarction and neurological deficits, even after 6 h of reperfusion. These findings suggest that inhibition of GAPDH aggregation may be a potential therapeutic target for AIS. Further efforts are warranted to translate these findings to treatment with AIS.
© 2025 The Author(s).
Product Citations: 462
Inhibition of GAPDH aggregation as a potential treatment for acute ischemic stroke.
In IScience on 20 June 2025 by Itakura, M., Kubo, T., et al.
-
Cardiovascular biology
Nesprin-2 contains BH3-like motifs that can promote cell death.
In Cell Death Discovery on 3 June 2025 by Zohar, H., Kessel, A., et al.
BH3-only proteins are a subgroup of the pro-apoptotic Bcl-2 family proteins. They initiate apoptosis by interacting with the multidomain pro- and anti-apoptotic Bcl-2 family proteins. SYNE2 encodes multiple nesprin-2 (Nes2) isoforms of which the most abundant and the largest is the nuclear envelope protein nesprin-2 giant (Nes2G). Nes2G is a component of the nuclear envelope Linker of Nucleoskeleton and Cytoskeleton (LINC) complex that connects the nucleus and the cytoskeleton. Previously, we showed that Nes2 has pro-apoptotic activity. We now show that Nes2G can bind multidomain pro-apoptotic and anti-apoptotic Bcl-2 family proteins and contains two BH3-like motifs near its N- and C-termini. Molecular modeling predicts that these BH3-like motifs have amphipathic α-helix structures and can dock onto the canonical groove of Bax and anti-apoptotic proteins as well as the trigger site of Bax. A chimeric tBid with its BH3 domain replaced with the C-terminal Nes2 BH3-like domain binds to Bax in cells. Furthermore, Nes2 BH3-like motif-containing fragments from the N- and the C-termini bind both pro-apoptotic and anti-apoptotic Bcl-2 proteins and promote cytochrome c release (indicative of apoptosis). Our results suggest that Nes2 acts as a BH3-only protein that regulates apoptosis by binding to the multidomain Bcl-2 family proteins.
© 2025. The Author(s).
-
Mus musculus (House mouse)
Oxidative Stress in Brain Function.
In Antioxidants (Basel, Switzerland) on 28 February 2025 by Trofin, D. M., Sardaru, D. P., et al.
Oxidative stress (OS) is an important factor in the pathophysiology of numerous neurodegenerative disorders, such as Parkinson's disease, multiple sclerosis, cerebrovascular pathology or Alzheimer's disease. OS also significantly influences progression among the various neurodegenerative disorders. The imbalance between the formation of reactive oxygen species (ROS) and the body's capacity to neutralize these toxic byproducts renders the brain susceptible to oxidative injury. Increased amounts of ROS can result in cellular malfunction, apoptosis and neurodegeneration. They also represent a substantial factor in mitochondrial dysfunction, a defining characteristic of neurodegenerative disorders. Comprehending the fundamental mechanisms of OS and its interactions with mitochondrial function, neuroinflammation and cellular protective pathways becomes essential for formulating targeted therapeutics to maintain brain health and reduce the impacts of neurodegeneration. We address recent highlights on the role of OS in brain function in terms of significance for neuronal health and the progression of neurodegenerative disorders.
An inherited mtDNA mutation remodels inflammatory cytokine responses in macrophages andin vivo
Preprint on BioRxiv : the Preprint Server for Biology on 5 January 2025 by Marques, E., Burr, S. P., et al.
Impaired mitochondrial bioenergetics in macrophages can drive hyperinflammatory cytokine responses 1–6 , but whether this may also be caused by inherited mtDNA mutations is unknown. Here, we address this question using a multi-omic approach that integrates super-resolution imaging and metabolic analyses to profile macrophages from a mouse model of mitochondrial disease arising from a heteroplasmic mutation (m.5019A>G) in the mitochondrial tRNA for alanine 7 . These m.5019A>G macrophages exhibit defects in respiratory chain complexes and oxidative phosphorylation (OxPhos) due to decreased intra-mitochondrial translation. To adapt to this metabolic stress, mitochondrial fusion, reductive glutamine metabolism, and aerobic glycolysis are all increased. Upon inflammatory activation, type I interferon (IFN-I) release is enhanced, while the production of pro-inflammatory cytokines and oxylipins are restrained in m.5019A>G macrophages. Finally, an in vivo endotoxemia model using m.5019A>G mice reveal elevated IFN-I levels and sickness behaviour. In conclusion, our study identifies an unexpected imbalance in innate immune signalling in response to a pathogenic mtDNA mutation, with important implications for the progression of pathology in patients with mtDNA diseases 8 .
-
Immunology and Microbiology
In Nature Communications on 2 January 2025 by Haas, M., Cherfa, S., et al.
Our study explores the complex dynamics of the integrated stress response (ISR) axis, highlighting PIM2 kinase's critical role and its interaction with the BCL2 protein family, uncovering key mechanisms of cell survival and tumor progression. Elevated PIM2 expression, a marker of various cancers, often correlates with disease aggressiveness. Using a model of normal and malignant plasma cells, we show that inhibiting PIM2 kinase inhibits phosphorylated BAD production and activates ISR-mediated NOXA expression. This shift towards MCL1 dependence underscores the synergy achieved through combined PIM/MCL1 inhibition, driven largely by ISR-mediated NOXA expression. In mouse xenograft models, dual targeting of PIM2 and MCL1 effectively controls tumor growth-a response reversed by ISR-specific inhibition and upregulation of genes linked to tumor cell dissemination. This work elucidates the molecular intricacies of PIM2 inhibition and its implications for cancer therapy, especially in tumors with elevated PIM2 expression.
© 2024. The Author(s).
-
ICC-IF
-
Mus musculus (House mouse)
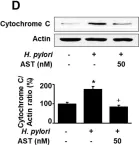
In Nutrients on 11 June 2020 by Lee, H., Lim, J. W., et al.
Fig.2.D

-
WB
-
Collected and cropped from Nutrients by CiteAb, provided under a CC-BY license
Image 1 of 10
In J Clin Biochem Nutr on 1 January 2020 by Takishita, Y., Yasuda, H., et al.
Fig.3.B

-
WB
-
Collected and cropped from J Clin Biochem Nutr by CiteAb, provided under a CC-BY license
Image 1 of 10
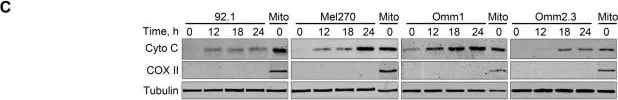
In Mol Cancer on 13 November 2019 by Zhou, J., Liu, S., et al.
Fig.2.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Mol Cancer by CiteAb, provided under a CC-BY license
Image 1 of 10
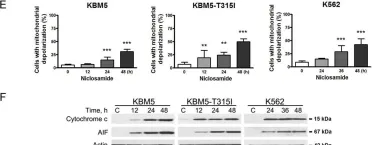
In Cell Death Dis on 22 January 2018 by Jin, B., Wang, C., et al.
Fig.4.E

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 10
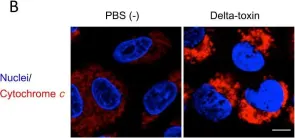
In PLoS One on 26 January 2016 by Seike, S., Miyamoto, K., et al.
Fig.7.B

-
ICC-IF
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 10
In Elife on 25 February 2014 by Yamano, K., Fogel, A. I., et al.
Fig.2.A

-
ICC-IF
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 10
In Elife on 25 February 2014 by Yamano, K., Fogel, A. I., et al.
Fig.2.B

-
ICC-IF
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 10
In Elife on 25 February 2014 by Yamano, K., Fogel, A. I., et al.
Fig.2.A

-
ICC-IF
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 10
In Elife on 25 February 2014 by Yamano, K., Fogel, A. I., et al.
Fig.2.B

-
ICC-IF
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 10
In Elife on 25 February 2014 by Yamano, K., Fogel, A. I., et al.
Fig.3.A

-
ICC-IF
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 10