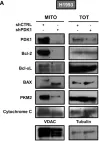
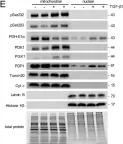
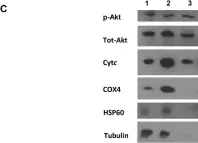
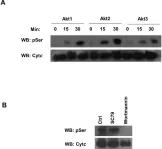
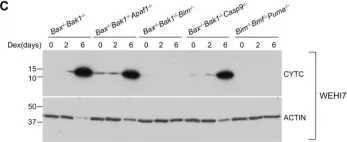
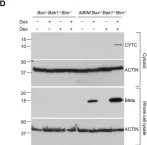
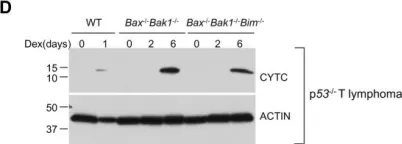
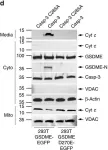
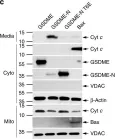
Ferroptosis and apoptosis are widely considered to be independent cell death modalities. Ferroptotic cell death is a consequence of insufficient radical detoxification and progressive lipid peroxidation, which is counteracted by glutathione peroxidase-4 (GPX4). Apoptotic cell death can be triggered by a wide variety of stresses, including oxygen radicals, and can be suppressed by anti-apoptotic members of the BCL-2 protein family. Mitochondria are the main interaction site of BCL-2 family members and likewise a major source of oxygen radical stress. We therefore studied if ferroptosis and apoptosis might intersect and possibly interfere with one another. Indeed, cells dying from impaired GPX4 activity displayed hallmarks of both ferroptotic and apoptotic cell death, with the latter including (transient) membrane blebbing, submaximal cytochrome-c release and caspase activation. Targeting BCL-2, MCL-1 or BCL-XL with BH3-mimetics under conditions of moderate ferroptotic stress in many cases synergistically enhanced overall cell death and frequently skewed primarily ferroptotic into apoptotic outcomes. Surprisingly though, in other cases BH3-mimetics, most notably the BCL-XL inhibitor WEHI-539, counter-intuitively suppressed cell death and promoted cell survival following GPX4 inhibition. Further studies revealed that most BH3-mimetics possess previously undescribed antioxidant activities that counteract ferroptotic cell death at commonly employed concentration ranges. Our results therefore show that ferroptosis and apoptosis can intersect. We also show that combining ferroptotic stress with BH3-mimetics, context-dependently can either enhance and convert cell death outcomes between ferroptosis and apoptosis or can also suppress cell death by intrinsic antioxidant activities.
© 2025. The Author(s).
Product Citations: 792
Interplay of ferroptotic and apoptotic cell death and its modulation by BH3-mimetics.
In Cell Death and Differentiation on 29 April 2025 by Qiu, Y., Hüther, J. A., et al.
-
Cell Biology
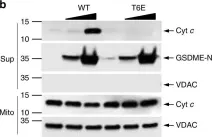
In Cell Death and Differentiation on 9 April 2025 by Gong, J., Djajawi, T. M., et al.
BH3 mimetic drugs that selectively target the pro-survival BCL2 proteins are highly promising for cancer treatment, most notably for treating blood cancers. Venetoclax, which inhibits BCL2, is now approved for treating chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML). Preferably, robust and validated assays would identify patients most likely to benefit from therapy with venetoclax itself or with inhibitors of other pro-survival proteins. A sophisticated method that has been developed is the BH3 profiling assay. In this assay, permeabilized, instead of intact, cells are treated for a few hours with inhibitors of the pro-survival BCL2 proteins, and the resultant mitochondrial depolarization measured. Sensitivity to a specific inhibitor (e.g., venetoclax or other BH3 mimetics) is then used to infer the reliance of a tumor (e.g., CLL) on one or more pro-survival BCL2 proteins. However, we found that this methodology cannot reliably identify such dependencies. In part, this is because almost all cells express multiple pro-survival BCL2 proteins that restrain BAX and BAK which must be inhibited before mitochondrial depolarization and apoptosis can proceed. Using genetic and pharmacological tools across multiple cell line models of blood cancer, we demonstrated that selective BCL2 inhibitors have important flow-on effects that includes the redistribution of BH3-only proteins to ancillary pro-survival proteins not directly engaged by the inhibitor. These secondary effects, critical to the biological action of selective inhibitors, were not accurately recapitulated in permeabilized cells, probably due to the limited time frame possible in such assays or the altered biophysical conditions when cells are permeabilized. While we could consistently define the sensitivity of a tumor cell to a particular BH3 mimetic drugs using intact cells, this was not reliable with permeabilized cells. These studies emphasize the need to carefully evaluate assays on permeabilized cells undertaken with inhibitors of the pro-survival BCL2 proteins.
© 2025. Crown.
-
Cancer Research
-
Cardiovascular biology
-
Cell Biology
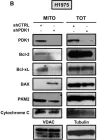
In Nature Communications on 11 March 2025 by Naghdi, S., Mishra, P., et al.
Differences between normal tissues and invading tumors that allow tumor targeting while saving normal tissue are much sought after. Here we show that scarcity of VDAC2, and the consequent lack of Bak recruitment to mitochondria, renders hepatocyte mitochondria resistant to permeabilization by truncated Bid (tBid), a Bcl-2 Homology 3 (BH3)-only, Bcl-2 family protein. Increased VDAC2 and Bak is found in most human liver cancers and mitochondria from tumors and hepatic cancer cell lines exhibit VDAC2- and Bak-dependent tBid sensitivity. Exploring potential therapeutic targeting, we find that combinations of activators of the tBid pathway with inhibitors of the Bcl-2 family proteins that suppress Bak activation enhance VDAC2-dependent death of hepatocarcinoma cells with little effect on normal hepatocytes. Furthermore, in vivo, combination of S63845, a selective Mcl-1 inhibitor, with tumor-nectrosis factor-related, apoptosis-induncing ligand (TRAIL) peptide reduces tumor growth, but only in tumors expressing VDAC2. Thus, we describe mitochondrial molecular fingerprint that discriminates liver from hepatocarcinoma and allows sparing normal tissue while targeting tumors.
© 2025. The Author(s).
-
WB
-
Homo sapiens (Human)
-
Rattus norvegicus (Rat)
-
Cancer Research
-
Cell Biology
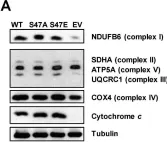
Cardiac Tyrosine 97 Phosphorylation of Cytochrome c Regulates Respiration and Apoptosis.
In International Journal of Molecular Sciences on 4 February 2025 by Morse, P. T., Pasupathi, V., et al.
It was previously reported that tyrosine 97 (Y97) of cytochrome c is phosphorylated in cow heart tissue under physiological conditions. Y97 phosphorylation was shown to partially inhibit respiration in vitro in the reaction with purified cytochrome c oxidase. Here, we use phosphomimetic Y97E Cytc to further characterize the functional effects of this modification both in vitro and in cell culture models. In vitro, phosphomimetic Y97E Cytc showed lower activity in the reaction with purified cow heart cytochrome c oxidase (COX), decreased caspase-3 activity, and reduced rate of reduction. Additionally, the phosphomimetic Y97E Cytc tended to be resistant to heme degradation and showed an increased rate of oxidation. Intact mouse Cytc double knockout fibroblasts were transfected with plasmids coding for phosphomimetic Y97E Cytc and other variants. Compared to cells expressing wild-type Cytc, the cells expressing phosphomimetic Y97E Cytc showed reduced respiration, mitochondrial membrane potential, and reactive oxygen species production, and protection from apoptosis. In an oxygen-glucose deprivation/reoxygenation cell culture model of ischemia/reperfusion injury, mitochondrial membrane potential and reactive oxygen species production were decreased. These data show that Cytc phosphorylation controls the overall flux through the electron transport chain by maintaining optimal intermediate ΔΨm potentials for efficient ATP production while minimizing reactive oxygen species production, thus protecting the cell from apoptosis.
-
WB
-
Mus musculus (House mouse)
-
Cardiovascular biology
Ferroptosis triggers mitochondrial fragmentation via Drp1 activation.
In Cell Death & Disease on 25 January 2025 by Pedrera, L., Prieto Clemente, L., et al.
Constitutive mitochondrial dynamics ensure quality control and metabolic fitness of cells, and their dysregulation has been implicated in various human diseases. The large GTPase Dynamin-related protein 1 (Drp1) is intimately involved in mediating constitutive mitochondrial fission and has been implicated in mitochondrial cell death pathways. During ferroptosis, a recently identified type of regulated necrosis driven by excessive lipid peroxidation, mitochondrial fragmentation has been observed. Yet, how this is regulated and whether it is involved in ferroptotic cell death has remained unexplored. Here, we provide evidence that Drp1 is activated upon experimental induction of ferroptosis and promotes cell death execution and mitochondrial fragmentation. Using time-lapse microscopy, we found that ferroptosis induced mitochondrial fragmentation and loss of mitochondrial membrane potential, but not mitochondrial outer membrane permeabilization. Importantly, Drp1 accelerated ferroptotic cell death kinetics. Notably, this function was mediated by the regulation of mitochondrial dynamics, as overexpression of Mitofusin 2 phenocopied the effect of Drp1 deficiency in delaying ferroptosis cell death kinetics. Mechanistically, we found that Drp1 is phosphorylated and activated after induction of ferroptosis and that it translocates to mitochondria. Further activation at mitochondria through the phosphatase PGAM5 promoted ferroptotic cell death. Remarkably, Drp1 depletion delayed mitochondrial and plasma membrane lipid peroxidation. These data provide evidence for a functional role of Drp1 activation and mitochondrial fragmentation in the acceleration of ferroptotic cell death, with important implications for targeting mitochondrial dynamics in diseases associated with ferroptosis.
© 2025. The Author(s).
-
Cell Biology
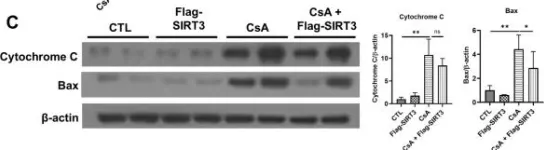
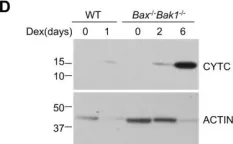
In Sci Rep on 2 May 2024 by Kim, J. E., Jo, M. J., et al.
Fig.3.C

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 56
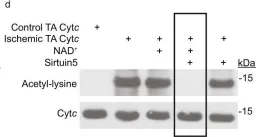
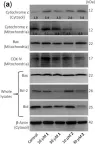
In Nat Commun on 13 July 2023 by Morse, P. T., Pérez-Mejías, G., et al.
Fig.1.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
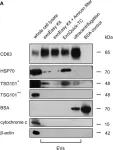
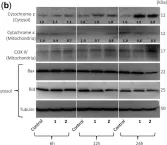
In Front Oncol on 21 October 2022 by Lang, J. B., Buck, M. C., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 56
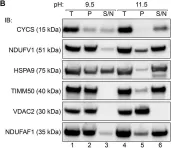
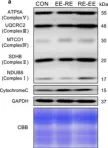
In Front Cell Dev Biol on 19 March 2022 by Robinson, D. R. L., Hock, D. H., et al.
Fig.1.B

-
WB
-
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 56
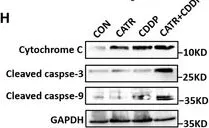
In EMBO Mol Med on 7 December 2021 by Zhao, L., Deng, X., et al.
Fig.6.H

-
WB
-
Collected and cropped from EMBO Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 56
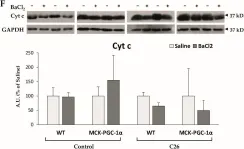
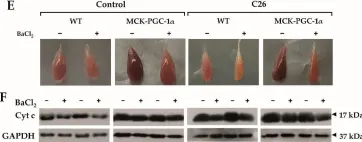
In Cells on 12 November 2021 by Beltrà, M., Pin, F., et al.
Fig.3.F

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cells on 12 November 2021 by Beltrà, M., Pin, F., et al.
Fig.3.E

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cancers (Basel) on 17 August 2021 by De Rosa, V., Iommelli, F., et al.
Fig.5.B

-
WB
-
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cancers (Basel) on 17 August 2021 by De Rosa, V., Iommelli, F., et al.
Fig.5.A

-
WB
-
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 56
In Sci Rep on 21 October 2020 by Smith, E. R. & Hewitson, T. D.
Fig.4.E

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cells on 6 August 2020 by Kalpage, H. A., Wan, J., et al.
Fig.6.C

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cells on 6 August 2020 by Kalpage, H. A., Wan, J., et al.
Fig.6.A

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cells on 6 August 2020 by Kalpage, H. A., Wan, J., et al.
Fig.1.A

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cell Death Dis on 8 June 2020 by Dong, L. & Vaux, D. L.
Fig.5.C

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cell Death Dis on 8 June 2020 by Dong, L. & Vaux, D. L.
Fig.6.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cell Death Dis on 8 June 2020 by Dong, L. & Vaux, D. L.
Fig.5.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 56
In Cell Death Dis on 8 June 2020 by Dong, L. & Vaux, D. L.
Fig.2.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 56
In Int J Mol Sci on 16 April 2020 by Saavedra, E., Estévez-Sarmiento, F., et al.
Fig.4.A

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 56
In Int J Mol Sci on 16 April 2020 by Saavedra, E., Estévez-Sarmiento, F., et al.
Fig.4.B

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 56
In Physiol Rep on 1 April 2020 by Shirai, T., Aoki, Y., et al.
Fig.4.A

-
WB
-
Collected and cropped from Physiol Rep by CiteAb, provided under a CC-BY license
Image 1 of 56
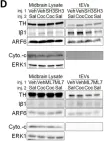
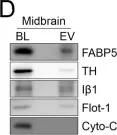
In Elife on 9 October 2019 by Nakamura, Y., Dryanovski, D. I., et al.
Fig.4.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 56
In Elife on 9 October 2019 by Nakamura, Y., Dryanovski, D. I., et al.
Fig.5.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 11 April 2019 by Rogers, C., Erkes, D. A., et al.
Fig.5.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 11 April 2019 by Rogers, C., Erkes, D. A., et al.
Fig.5.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56
In Nat Commun on 11 April 2019 by Rogers, C., Erkes, D. A., et al.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 56