
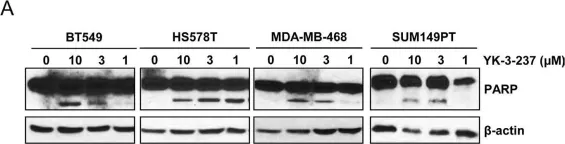
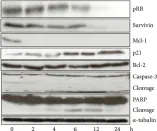
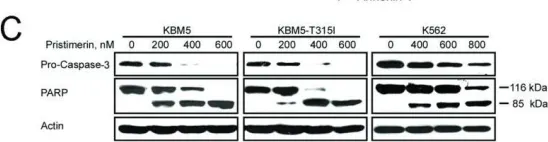
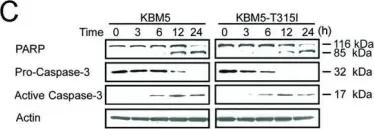
Prostate cancer (PCa) is the second most common cancer worldwide and the fifth leading cause of cancer-related deaths among men. The emergence of metastatic castration-resistant prostate cancer (mCRPC) after androgen deprivation therapy (ADT) exemplifies the complex disease management for PCa. PARP inhibitors (PARPis) are being tested to treat mCRPC in tumors with defective homologous recombination repair (HRR) to address this complexity. However, increasing resistance towards PARPi in HRR-deficient patients and the low percentage of HRR-defective mCRPC patients requires the identification of new genes whose deficiency can be exploited for PARPi treatment. XRCC1 is a DNA repair protein critical in the base excision repair (BER) and single strand break repair (SSBR) pathways. We analyzed PCa patients' cohorts and found that XRCC1 expression varies widely, with many patients showing low XRCC1 expression. We created XRCC1 deficiency in PCa models to examine PARPi sensitivity. XRCC1 loss conferred hypersensitivity to PARPi by promoting the accumulation of DNA double-strand breaks, increasing cell-cycle arrest, and inducing apoptosis. We confirmed that XRCC1 expression correlated with PARPi sensitivity using a doxycycline-inducible system. Therefore, we conclude that XRCC1 expression level predicts response to PARPi, and the clinical utility of PARPi in PCa can extend to low XRCC1 expressing tumors.
© The Author(s) 2025. Published by Oxford University Press on behalf of NAR Cancer.
Product Citations: 142
PARP inhibitor response is enhanced in prostate cancer when XRCC1 expression is reduced.
In NAR Cancer on 1 June 2025 by Goel, K., Venkatappa, V., et al.
-
Cancer Research
Interplay of ferroptotic and apoptotic cell death and its modulation by BH3-mimetics.
In Cell Death and Differentiation on 29 April 2025 by Qiu, Y., Hüther, J. A., et al.
Ferroptosis and apoptosis are widely considered to be independent cell death modalities. Ferroptotic cell death is a consequence of insufficient radical detoxification and progressive lipid peroxidation, which is counteracted by glutathione peroxidase-4 (GPX4). Apoptotic cell death can be triggered by a wide variety of stresses, including oxygen radicals, and can be suppressed by anti-apoptotic members of the BCL-2 protein family. Mitochondria are the main interaction site of BCL-2 family members and likewise a major source of oxygen radical stress. We therefore studied if ferroptosis and apoptosis might intersect and possibly interfere with one another. Indeed, cells dying from impaired GPX4 activity displayed hallmarks of both ferroptotic and apoptotic cell death, with the latter including (transient) membrane blebbing, submaximal cytochrome-c release and caspase activation. Targeting BCL-2, MCL-1 or BCL-XL with BH3-mimetics under conditions of moderate ferroptotic stress in many cases synergistically enhanced overall cell death and frequently skewed primarily ferroptotic into apoptotic outcomes. Surprisingly though, in other cases BH3-mimetics, most notably the BCL-XL inhibitor WEHI-539, counter-intuitively suppressed cell death and promoted cell survival following GPX4 inhibition. Further studies revealed that most BH3-mimetics possess previously undescribed antioxidant activities that counteract ferroptotic cell death at commonly employed concentration ranges. Our results therefore show that ferroptosis and apoptosis can intersect. We also show that combining ferroptotic stress with BH3-mimetics, context-dependently can either enhance and convert cell death outcomes between ferroptosis and apoptosis or can also suppress cell death by intrinsic antioxidant activities.
© 2025. The Author(s).
-
Cell Biology
In Cancer Research and Treatment : Official Journal of Korean Cancer Association on 7 April 2025 by Kim, M., Min, A., et al.
Sustained cell proliferation and cell cycle acceleration in cancer cells inherently increase DNA damage, which interferes with homeostatic replication and transcription. Ataxia telangiectasia and Rad3-related (ATR) is crucial for initiation of the DNA damage response, and ATR inhibitors, such as elimusertib, induce increased replication stress and DNA damage. We investigated the anti-tumor effects of elimusertib and its mechanism of action in relation to replication stress.
Anti-tumor effects were evaluated by MTT assay and colony formation assay in breast cancer cell lines in vitro, in breast cancer cell xenografts in vivo, and in patient-derived xenograft models. Cell cycle was assessed by flow cytometry and BrdU assay was used to measure replicating cells and S-phase progression. Alkaline and neutral comet assay was used to measure single and double stranded DNA damages, respectively.
Elimusertib delayed S-phase progression in MDA-MB-453 and MDA-MB-231 cells and induced caspase-7-dependent apoptosis. Furthermore, the increase in sub-G1 population in the FACS analysis and Annexin V assay also confirmed apoptotic cell death. In the BrdU assay, single stranded DNA (ssDNA) increased in sensitive cells and aberrant ssDNA induced DNA damage in S-phase and eventually caused replication catastrophe. Finally, these anti-tumor effects were proven in in vivo xenograft and patient-derived xenograft models.
Elimusertib had anti-tumor effects and induced replication catastrophe in breast cancer cells with a high replication rate. Moreover, cells under high DNA replication stress were sensitive to elimusertib. Further studies and treatment strategies with elimusertib are warranted for cancers with a high replication rate.
-
Cancer Research
In Frontiers in Pharmacology on 3 April 2025 by Nizami, Z. N., Al Azzani, M., et al.
Colorectal cancer is a leading cause of cancer related-death worldwide, and resistance to 5-fluorouracil (5FU, a key component of chemotherapy regimens, is a major clinical concern. We have previously elucidated the effects of Rhus coriaria ethanolic extract (RCE) in triple-negative breast cancer, CRC, and pancreatic cancer cells. Here, we explored the anticancer effects of RCE in parental (HCT-116-WT) and 5FU-resistant HCT-116 (HCT-116-5FU-R) CRC cells.
MTT assay was used to assess cell viability. Muse analyzer was used to assess cell viability, cell cycle distribution, and apoptosis. Additionally, colony formation and growth assays and western blots were performed. In vivo effects of RCE were assessed by an in ovo chick embryo tumor growth assay.
We found that RCE inhibited the viability and colony formation and growth capacities of HCT-116-WT and HCT-116-5FU-R cells. The antiproliferative effects were attributed to DNA damage-mediated impairment of cell cycle at S phase, and induction of Beclin-1-independent autophagy in both cell lines. Mechanistically, inhibition of the mTOR, STAT3 and p38 MAPK pathways was implicated in the latter. Additionally, RCE induced caspase-7-independent apoptosis in HCT-116-WT cells. However, HCT-116-5FU-R cells were resistant to apoptosis through upregulation of survivin, and downregulation of Bax. Using autophagy and proteasome inhibitors, we clarified that autophagy and the proteasome pathway contributed to RCE-mediated cell death in HCT-116-WT and HCT-116-5FU-R cells. Lastly, we confirmed RCE inhibited the growth of both HCT-116-WT and HCT-116-5FU-R xenografts in a chick embryo model.
Collectively, our findings highlight that RCE is a source of phytochemicals that can be used as anticancer agents for 5FU-resistant CRC.
Copyright © 2025 Nizami, Al Azzani, Khaldi, Wali, Magramane, Samad, Eid, Arafat, Al Dhaheri, Attoub and Iratni.
-
Cancer Research
-
Pharmacology
TRAF2 and RIPK1 redundantly mediate classical NFκB signaling by TNFR1 and CD95-type death receptors.
In Cell Death & Disease on 21 January 2025 by Wagner, J., Vredevoogd, D., et al.
This study suggests a modified model of TNFR1-induced complex I-mediated NFκB signaling. Evaluation of a panel of five tumor cell lines (HCT116-PIK3CAmut, SK-MEL-23, HeLa-RIPK3, HT29, D10) with TRAF2 knockout revealed in two cell lines (HT29, HeLa-RIPK3) a sensitizing effect for death receptor-induced necroptosis and in one cell line (D10) a mild sensitization for TNFR1-induced apoptosis. TRAF2 deficiency inhibited death receptor-induced classical NFκB-mediated production of IL-8 only in a subset of cell lines and only partly. TRAF5, furthermore, failed to improve DR-induced NFκB signaling in HCT116-PIK3CAmut and HCT116-PIK3CAmut-TRAF2KO cells. These findings argue for a non-obligatory role of TRAF2 in death receptor-induced classical NFκB signaling. Similar as in TRAF2-deficient cells, TNF- and CD95L-induced NFκB signaling was found to be only poorly affected in RIPK1KO cells and in cells treated with the RIPK1-specific PROTAC LD4172. Intriguingly, however, death receptor-induced NFκB signaling was completely inhibited in HCT116-PIK3CAmut cells double deficient for TRAF2 and RIPK1 and in TRAF2-deficient cells treated with LD4172. Moreover, with exception of recruitment of TRADD, acting upstream to TRAF2 and parallel to RIPK1, TNFR1 signaling complex formation was abrogated in TRAF2-RIPK1 DKO cells. Based on our findings, two distinguishable types of TNFR1-interacting complexes promote TNF-induced NFκB signaling: First, a TRADD-TRAF2/cIAP utilizing complex Ia which becomes evident in RIPK1-deficient cells. Second, a non-modified RIPK1 utilizing complex Ib which acts in TRADD- or TRAF2-deficient cells. Complex Ia and Ib may furthermore interact and cooperate to ubiquitinate RIPK1 resulting in a modified complex Ia/b preventing complex Ia and Ib to convert to the established TNFR1-induced cytotoxic complexes IIa and IIb.
© 2025. The Author(s).
-
Homo sapiens (Human)
-
Cell Biology
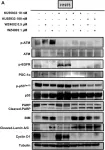
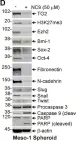
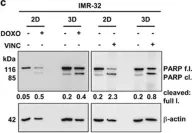
In Cancer Metab on 6 November 2023 by Terlizzi, C., De Rosa, V., et al.
Fig.4.A

-
WB
-
Collected and cropped from Cancer Metab by CiteAb, provided under a CC-BY license
Image 1 of 21
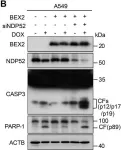
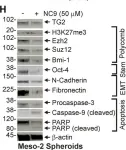
In Cancer Metab on 6 November 2023 by Terlizzi, C., De Rosa, V., et al.
Fig.5.A

-
WB
-
Collected and cropped from Cancer Metab by CiteAb, provided under a CC-BY license
Image 1 of 21
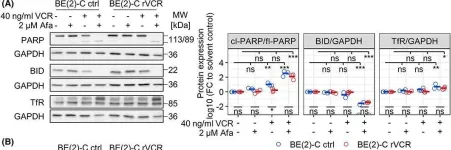
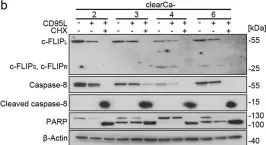
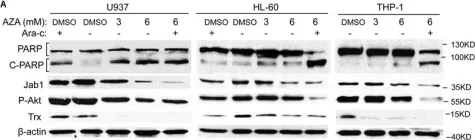
In Cell Death Dis on 30 September 2023 by Mu, N., Wang, Y., et al.
Fig.5.B

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 21
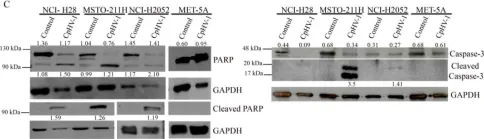
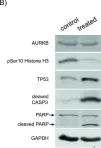
In Mol Oncol on 1 January 2023 by Rösch, L., Herter, S., et al.
Fig.7.A

-
WB
-
Collected and cropped from Mol Oncol by CiteAb, provided under a CC-BY license
Image 1 of 21
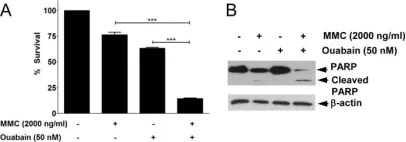
In Viruses on 8 December 2021 by Forte, I. M., Indovina, P., et al.
Fig.2.C

-
WB
-
Collected and cropped from Viruses by CiteAb, provided under a CC-BY license
Image 1 of 21
In Cell Death Dis on 16 May 2019 by Luebke, T., Schwarz, L., et al.
Fig.2.B

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 21
In Oncotarget on 2 October 2018 by Adhikary, G., Grun, D., et al.
Fig.4.D

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 21
In Oncotarget on 2 October 2018 by Adhikary, G., Grun, D., et al.
Fig.4.H

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 21
In J Virol on 1 July 2018 by Natesampillai, S., Cummins, N. W., et al.
Fig.4.C

-
WB
-
Collected and cropped from J Virol by CiteAb, provided under a CC-BY license
Image 1 of 21
In PLoS One on 9 May 2018 by Clarisse, D., Van Wesemael, K., et al.
Fig.1.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 21
In PLoS One on 9 May 2018 by Clarisse, D., Van Wesemael, K., et al.
Fig.3.D

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 21
In Cell Death Dis on 24 August 2017 by Bingel, C., Koeneke, E., et al.
Fig.7.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 21
In Cell Death Dis on 24 August 2017 by Bingel, C., Koeneke, E., et al.
Fig.7.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 21
In Cell Death Dis on 24 August 2017 by Bingel, C., Koeneke, E., et al.
Fig.3.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 21
In Front Pharmacol on 1 July 2017 by Pan, Y., Liu, D., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Pharmacol by CiteAb, provided under a CC-BY license
Image 1 of 21
In Oncotarget on 3 November 2015 by Bogen, D., Wei, J. S., et al.
Fig.6.B

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 21
In PLoS One on 15 October 2013 by Jun, D. W., Hwang, M., et al.
Fig.5.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 21
In Oncotarget on 1 July 2013 by Yi, Y. W., Kang, H. J., et al.
Fig.4.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 21
In J Anal Methods Chem on 12 April 2013 by Cui, C., Wang, Y., et al.
Fig.5.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Anal Methods Chem by CiteAb, provided under a CC-BY license
Image 1 of 21
In Mol Cancer on 19 May 2010 by Lu, Z., Jin, Y., et al.
Fig.7.C

-
WB
-
Collected and cropped from Mol Cancer by CiteAb, provided under a CC-BY license
Image 1 of 21
In Mol Cancer on 19 May 2010 by Lu, Z., Jin, Y., et al.
Fig.4.C

-
WB
-
Collected and cropped from Mol Cancer by CiteAb, provided under a CC-BY license
Image 1 of 21