

Aim: Immune checkpoint inhibitors (ICIs) have revolutionized the treatment approach for NSCLC. However, the effectiveness of ICI therapy in patients with EGFR-driven NSCLC, particularly those resistant to EGFR-TKI, has been disappointing. The immunosuppressive tumor microenvironment (TME) following EGFR-TKI therapy has been proved to significantly affected the effectiveness of ICIs. Therefore, studying the mechanism behind the development of a suppressive TME and exploring potential interventions is crucial for research on EGFR-TKI-resistant NSCLC. Methods: ZEB2 levels were quantified in human NSCLC cell lines and in tumor specimens from NSCLC patients by quantitative RT-PCR (qRT-PCR), WB, and immunohistochemical staining. To examine how ZEB2 affected macrophage polarization, M1/M2 marker profiles were measured with qRT-PCR and flow cytometry. Changes in cytokine production triggered by altered ZEB2 expression were determined with qRT-PCR, ELISA, and Meso Scale Discovery electrochemiluminescence assays. The direct binding of ZEB2 to cytokine-gene promoters was tested using a dual-luciferase reporter system. Upstream regulatory pathways were investigated by correlating LUAD transcriptomic data from TCGA with ZEB2 expression and validating key findings via western blotting. Finally, cell-derived xenograft (CDX) models were generated by subcutaneously implanting pre-treated PC9 or HCC827 cells into BALB/c nude mice to verify the impact of EGFR-TKI resistance and ZEB2 on tumor-associated macrophage (TAM) polarization in vivo. Results: It was elucidated that EGFR-TKI resistance upregulated the M2 polarization biomarkers, Arg-1 (PC9-GR: P < 0.01; HCC827-GR: P < 0.05) and IL4 (PC9-GR: P < 0.01; HCC827-GR: P < 0.01), while downregulated the M1 polarization biomarkers, TNF-α (PC9-GR: P < 0.01; HCC827-GR: P < 0.01), IL1β (PC9-GR: P < 0.01; HCC827-GR: P < 0.01), and IL6(PC9-GR: P < 0.001; HCC827-GR: P < 0.001) in NSCLC cell lines. Meanwhile, CD206+ TAMs (PC9-GR: P < 0.05; HCC827-GR: P < 0.01) were increased and CD86+ TAMs (PC9-GR: P < 0.05; HCC827-GR: P < 0.05) were decreased in both EGFR-TKI-resistant mice models. Apart from the formation of suppressive TME, ZEB2 was found to be upregulated in PC9-GR (qRT-PCR: P < 0.0001; WB: P < 0.05) and HCC827-GR (qRT-PCR: P < 0.0001; WB: P < 0.05) cells. The same trend was also noticed in clinical samples, with ZEB2 upregulated after gefitinib resistance in NSCLC patients (P < 0.0001). Based on these findings, ZEB2 knockdown was proved to downregulate Arg-1 (PC9-GR: P < 0.01; HCC827-GR: P < 0.05) and IL4 (PC9-GR: P < 0.01; HCC827-GR: P < 0.001), while upregulate the TNF-α (PC9-GR: P < 0.0001; HCC827-GR: P < 0.0001), IL1β (HCC827-GR: P < 0.001), and IL6 (PC9-GR: P < 0.01; HCC827-GR: P < 0.001), indicating its role in M1/M2 polarization in both EGFR-TKI-resistant NSCLC cell lines. The downregulation of CD206+ TAMs (PC9-GR: P < 0.05; HCC827-GR: P < 0.01) and the upregulation of CD86+ TAMs (PC9-GR: P < 0.001; HCC827-GR: P < 0.05) also demonstrated the reversion of suppressive TME after ZEB2 knockout in EGFR-TKI-resistant mice models. Additionally, after the intervention of MK2206, which was an Akt inhibitor, ZEB2 expression was suppressed at both low (PC9-GR: P < 0.001; HCC827-GR: P < 0.001) and high concentrations (PC9-GR: P < 0.001; HCC827-GR: P < 0.0001). Finally, the mechanism underlying ZEB2's regulation on TAM polarization was proved to be associated with cytokine secretion. According to the results of ELISA, apart from its inducement on TGF-β1 secretion (PC9-GR: P < 0.0001; HCC827-GR: P < 0.0001), ZEB2 could directly bind to the promoter region of CSF-1 to elevate its secretion (PC9-GR: P < 0.0001; HCC827-GR: P < 0.0001). Conclusion: In EGFR-TKI-resistant NSCLC, activation of the PI3K-Akt cascade drove a marked rise in ZEB2 expression. The elevated ZEB2 increased CSF-1 and TGF-β1 release, steering macrophages toward an M2 phenotype while impeding M1 polarization. Accordingly, suppressing ZEB2 had the potential to reshape the TME and enhance the effectiveness of ICIs once EGFR-TKI resistance had emerged.
© The Author(s) 2025.
Product Citations: 182
In Cancer Drug Resistance (Alhambra, Calif.) on 13 June 2025 by Liu, Y., Yu, Y., et al.
In Diabetes Metabolism Journal on 22 May 2025 by Song, D. G., Pak, S., et al.
Obesity is a rapidly increasing global health issue, which is associated with glucose and insulin resistance. Phosphodiesterase type 5 (PDE5) inhibitors (PDE5i) are known for their ability to enhance blood flow and vascular stability and are widely used to treat conditions such as erectile dysfunction, pulmonary hypertension, heart failure, and cancer. However, studies investigating the role of PDE5i in alleviating obesity and metabolic diseases remains unclear. Therefore, we investigated the effects of PDE5i on obesity and metabolic disorders in diet-induced obese mice and its underlying mechanisms.
PDE5i was administered to high-fat diet (HFD)-fed C57BL/6J mice for 6 to 7 weeks. Body weight and food intake were measured weekly, and baseline metabolic rates, physical activity, and glucose and insulin tolerance tests were assessed during PDE5i administration. Macrophages and T-cells in the gonadal white adipose tissue (gWAT) were analyzed by flow cytometry. Vascular stability and blood flow in gWAT were analyzed via immunostaining and in vivo live imaging. RAW264.7 cells and bone marrow-derived macrophages were used to determine immunoregulatory effects of PDE5i.
In HFD-fed mice, PDE5i administration significantly enhanced systemic insulin sensitivity and AKT phosphorylation in gWAT. PDE5i reduced the M1/M2 ratio of gWAT macrophages of obese mice. These phenomena were associated with enhanced blood flow to the gWAT. In vitro experiments revealed that PDE5i suppressed lipopolysaccharide-induced proinflammatory cytokine production and increased the mRNA expression of genes associated with M2 polarization.
PDE5i plays a role in regulating adipose tissue inflammation and thus holds promise as a therapeutic agent for metabolic enhancement.
-
Endocrinology and Physiology
-
Immunology and Microbiology
In International Journal of Biological Sciences on 19 May 2025 by Huo, S., Wang, M., et al.
The macrophage-cardiomyocyte crosstalk as a potential intervention target for diabetic cardiomyopathy (DCM) remains deeper exploration. We found S100A9, as an immunoinflammatory mediator, was up-regulated in cardiomyocytes and macrophages in diabetic heart by single-cell analysis. Furthermore, F4/80+CCR2+S100A9+ macrophages in peripheral blood and heart both increased in diabetic mice. S100A9 blocking by paquinimod or macrophage depletion (clodronate) alleviated diabetes-induced cardiac dysfunction, inflammatory macrophage infiltration, serum pro-inflammatory cytokines. More importantly, diabetic cardiac dysfunction, myocardial remodeling, and inflammation could be suppressed by macrophage specific S100A9 knockout (S100a9flox/floxLyz2-Cre). S100A9 activation led to excessive mitochondrial fission, decreased mitophagy flux, and elevated mitochondrial oxidative stress. In addition, proteomics and transcription factor profiling array unveiled S100A9 activated STAT3 in cardiomyocytes. Nevertheless, these effects were mitigated by STAT3(Y705F) mutation, STAT3 knockdown, or paquinimod. Our study highlights macrophage-derived S100A9 as a critical mediator for impaired mitochondrial quality control in diabetic cardiac dysfunction, and targeting S100A9 represents a promising therapeutic target.
© The author(s).
-
Cardiovascular biology
-
Cell Biology
-
Immunology and Microbiology
USP2 inhibition unleashes CD47-restrained phagocytosis and enhances anti-tumor immunity.
In Nature Communications on 16 May 2025 by Dai, P., Sun, Y., et al.
The CD47/SIRPα axis conveys a 'don't eat me' signal, thereby thwarting the phagocytic clearance of tumor cells. Although blocking antibodies targeting CD47 have demonstrated promising anti-tumor effects in preclinical models, clinical trials involving human cancer patients have not yielded ideal results. Exploring the regulatory mechanisms of CD47 is imperative for devising more efficacious combinational therapies. Here, we report that inhibiting USP2 prompts CD47 degradation and reshapes the tumor microenvironment (TME), thereby enhancing anti-PD-1 immunotherapy. Mechanistically, USP2 interacts with CD47, stabilizing it through deubiquitination. USP2 inhibition destabilizes CD47, thereby boosting macrophage phagocytosis. Single-cell RNA sequencing shows USP2 inhibition reprograms TME, evidenced by increasing M1 macrophages and CD8+ T cells while reducing M2 macrophages. Combining ML364 with anti-PD-1 reduces tumor burden in mouse models. Clinically, low USP2 expression predicts a better response to anti-PD-1 treatment. Our findings uncover the regulatory mechanism of CD47 by USP2 and targeting this axis boosts anti-tumor immunity.
© 2025. The Author(s).
-
Cancer Research
-
Immunology and Microbiology
Macrophage PKM2 depletion ameliorates hepatic inflammation and acute liver injury in mice.
In Frontiers in Pharmacology on 12 May 2025 by Kang, Z., Xie, R., et al.
Pyruvate kinase M2 (PKM2), the rate-limiting enzyme of glycolysis, plays a critical role in macrophage activation and a broad spectrum of chronic liver diseases. However, whether PKM2 contributes to the pathogenesis of acute liver injury (ALI) remains largely unexplored.
PKM2 expression was assessed in human and mouse ALI livers. Macrophage-specific PKM2 knockout mice were challenged by two independent ALI models, induced by acetaminophen (APAP) and lipopolysaccharide/D-galactosamine (LPS/D-GalN), to explore the role and regulatory mechanism of macrophage PKM2 in ALI progression.
By bioinformatic screening and analysis of ALI liver, we found that PKM2 was significantly upregulated in the liver tissues of ALI patients and mice. Immunofluorescence staining further demonstrated that PKM2 was markedly upregulated in macrophages during ALI progression. Notably, macrophage PKM2 depletion effectively alleviated APAP- and LPS/D-GalN-induced ALI, as demonstrated by ameliorated immune cells infiltration, pro-inflammatory mediators, and hepatocellular cell death. PKM2-deficient macrophages showed M2 anti-inflammatory polarization in vivo and in vitro. Furthermore, PKM2 deletion limited HIF-1α signaling and aerobic glycolysis of macrophages, which thereby attenuated macrophage pro-inflammatory activation and hepatocyte injury. Pharmacological PKM2 antagonist efficiently ameliorated liver injury and prolonged the survival of mice in APAP-induced ALI model.
Our study highlights the pivotal role of macrophage PKM2 in advancing ALI, and therapeutic targeting of PKM2 may serve as a novel strategy to combat ALI.
Copyright © 2025 Kang, Xie, Cui, Chen, Xie, Li, Lv, Ye, Zhao, Zhang, Hong and Qu.
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
-
Pharmacology
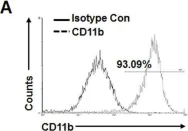
In EMBO Mol Med on 7 October 2021 by Bernasconi, R., Thriene, K., et al.
Fig.2.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from EMBO Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 4
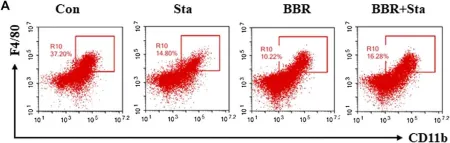
In Front Pharmacol on 17 November 2020 by Cao, H., Li, C., et al.
Fig.4.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Front Pharmacol by CiteAb, provided under a CC-BY license
Image 1 of 4
In Sci Rep on 11 September 2017 by Kim, S. M., Mun, B. R., et al.
Fig.4.A

-
FC/FACS
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 4
In PLoS One on 20 April 2014 by Sheng, K. C., Herrero, L. J., et al.
Fig.1.A

-
FC/FACS
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 4