

Psoriasis is a chronic inflammatory skin disease characterised by inflammatory cell infiltration, keratinocyte hyperproliferation and increased neovascularization. Despite extensive research, the precise mechanisms underlying psoriasis pathology and treatment strategies remain unclear because of a complex aetiology and disease progression. Hence, in this study, we aimed to identify potential therapeutic targets for psoriasis and explore their effects on disease progression. We observed that G protein-coupled receptor LGR4 attenuates psoriasis progression. Bioinformatics analysis of publicly available clinical data revealed lower LGR4 expression in the skin lesions of patients with psoriasis than in their non-lesioned skin. Both in vitro (HaCaT cell) and in vivo (mouse) models confirmed this phenomenon. The Lgr4-knockout mouse model further confirmed that LGR4 plays a positive role in psoriasis progression. Specifically, Lgr4 knockout promoted the secretion of inflammatory factors, accumulation of local immunocyte infiltration in skin lesions, and keratinocyte proliferation. In conclusion, we demonstrated that LGR4 is critical to limiting psoriasis progression, suggesting that it is a viable target for the clinical management of this skin condition.
© 2024 John Wiley & Sons Ltd.
Product Citations: 73
In Immunology on 1 February 2025 by Xue, M., Yang, R., et al.
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
Genetically engineered cellular nanoparticles loaded with curcuminoids for cancer immunotherapy.
In Theranostics on 21 October 2024 by Liao, Y., Zhao, C., et al.
Background: Inducing immunogenic cell death (ICD) is a promising strategy to enhance immune responses for immune checkpoint blockade (ICB) therapy, but the lack of a simple and effective platform to integrate ICD and ICB therapy limits their clinical application. Methods: Here, we developed programmed cell death protein 1 (PD1)-overexpressing genetically engineered nanovesicles (NVs)-coated curcumin (Cur)-loaded poly (lactic-co-poly-polyglycolic acid) nanoparticles (PD1@Cur-PLGA) to integrate ICD and ICB therapy for enhancing tumor immunotherapy. Results: Genetically engineered NVs greatly enhanced the tumor targeting of nanoparticles, and the PD1 on NVs dramatically blocked the PD1/PDL1 signaling pathway and stimulated antitumor immune responses. Meanwhile, the delivered Cur successfully induced tumor cell apoptosis and activated ICD by inhibiting NF-κB phosphorylation and Bcl-2 protein expression and activating caspase and Bax apoptotic signaling. By synergizing the ICD effect of Cur and the PD1/PDL1 axis blocking function of genetically engineered NVs, the PD1@Cur-PLGA enhanced the intratumoral infiltration rate of mature dendritic cells and CD8+ T cells in tumor tissues, resulting in significantly inhibiting tumor growth in breast and prostate tumor-bearing mouse models. Conclusion: This synergistic ICD and ICB therapy based on genetically engineered NVs provides a low-cost, safe, and effective strategy to enhance cancer immunotherapy.
© The author(s).
-
Mus musculus (House mouse)
-
Cancer Research
-
Immunology and Microbiology
In Journal of Nanobiotechnology on 12 September 2024 by Zeng, B., Pian, L., et al.
Breast cancer therapy has significantly advanced by targeting the programmed cell death-ligand 1/programmed cell death-1 (PD-L1/PD-1) pathway. BMS-202 (a smallmolecule PD-L1 inhibitor) induces PD-L1 dimerization to block PD-1/PD-L1 interactions, allowing the T-cell-mediated immune response to kill tumor cells. However, immunotherapy alone has limited effects. Clinically approved photodynamic therapy (PDT) activates immunity and selectively targets malignant cells. However, PDT aggravates hypoxia, which may compromise its therapeutic efficacy and promote tumor metastasis. We designed a tumor-specific delivery nanoplatform of liposomes that encapsulate the hypoxia-sensitive antitumor drug tirapazamine (TPZ) and the small-molecule immunosuppressant BMS. New indocyanine green (IR820)-loaded polyethylenimine-folic acid (PEI-FA) was complexed with TPZ and BMS-loaded liposomes via electrostatic interactions to form lipid nanocomposites. This nanoplatform can be triggered by near-infrared irradiation to induce PDT, resulting in a hypoxic tumor environment and activation of the prodrug TPZ to achieve efficient chemotherapy. The in vitro and in vivo studies demonstrated excellent combined PDT, chemotherapy, and immunotherapy effects on the regression of distant tumors and lung metastases, providing a reference method for the preparation of targeted agents for treating breast cancer.
© 2024. The Author(s).
-
Mus musculus (House mouse)
-
Cancer Research
-
Immunology and Microbiology
In Translational Cancer Research on 30 June 2024 by Zhu, X., Zhang, W., et al.
Emerging evidence suggests that immunogenic chemotherapy not only kills tumor cells but also improves the immune-suppressive tumor microenvironment by inducing immunogenic cell death (ICD), leading to sustained anti-tumor effects. The lack of ICD inducers explored in lung cancer necessitates investigation into new inducers for this context, therefore, this study aims to explore whether the gemcitabine (GEM) and celecoxib can activate the immunogenic chemotherapy progress in lung cancer tissue.
We assessed five chemotherapeutic agents for their ability to trigger ICD using ex vivo and in vivo experiments, including western blotting (WB), flow cytometry, and tumor preventive vaccine assays. Additionally, we evaluated the synergistic effects of GEM, celecoxib, and anti-programmed death 1 monoclonal antibody (aPD-1) in tumor-bearing mice to understand how GEM activates antitumor immunity and enhances immunochemotherapy.
GEM was identified as an effective ICD inducer, showing high expression of calreticulin (CRT) and heat shock protein 90 (HSP90). Co-culture with GEM-treated cells [Lewis lung carcinoma (LLC) and CMT-64] enhanced dendritic cell (DC) activity, evidenced by maturation markers and increased phagocytic capacity. Moreover, celecoxib was found to enhance ICD by reducing indoleamine 2,3-dioxygenase 1 (IDO1) expression and increasing reactive oxygen species (ROS)-based endoplasmic reticulum (ER) stress. The combination therapy [GEM, celecoxib, and aPD-1 (GCP)] exhibited potent and sustained antitumor activity in immunocompetent mice, with enhanced recruitment of tumor-infiltrating lymphocytes.
These findings support the potential use of GCP therapy as a treatment option for lung cancer patients.
2024 Translational Cancer Research. All rights reserved.
-
FC/FACS
In Diabetes, Obesity Metabolism on 1 June 2024 by Iwamoto, Y., Kimura, T., et al.
Dipeptidyl peptidase-4 (DPP-4) inhibitors suppress the inactivation of incretin hormones and lower blood glucose levels by inhibiting DPP-4 function. Sodium-glucose cotransporter 2 (SGLT2) inhibitors lower blood glucose levels in an insulin-independent manner by inhibiting renal reabsorption of glucose. DPP-4 and SGLT2 inhibitors each have the potential to improve hepatic steatosis; however, their combined effects remain unclear. In this study, we examined the effects of the combination of these drugs on hepatic steatosis using high-fat diet-fed mice.
C57BL/6J male mice were fed a 60% high-fat diet for 2 months to induce hepatic steatosis. Mice were divided into four groups (control; DPP-4 inhibitor anagliptin; SGLT2 inhibitor luseogliflozin; anagliptin and luseogliflozin combination), and the effects of each drug and their combination on hepatic steatosis after a 4-week intervention were evaluated.
There were no differences in blood glucose levels among the four groups. Anagliptin suppresses inflammation- and chemokine-related gene expression. It also improved macrophage fractionation in the liver. Luseogliflozin reduced body weight, hepatic gluconeogenesis and blood glucose levels in the oral glucose tolerance test. The combination treatment improved hepatic steatosis without interfering with the effects of anagliptin and luseogliflozin, respectively, and fat content and inflammatory gene expression in the liver were significantly improved in the combination group compared with the other groups.
The combination therapy with the DPP-4 inhibitor anagliptin and the SGLT2 inhibitor luseogliflozin inhibits fat deposition in the liver via anti-inflammatory effects during the early phase of diet-induced liver steatosis.
© 2024 John Wiley & Sons Ltd.
-
FC/FACS
-
Mus musculus (House mouse)
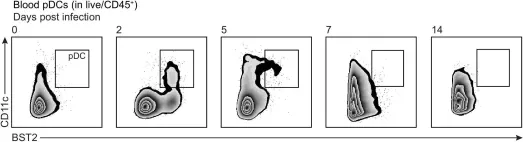
In Elife on 13 January 2022 by Gawish, R., Starkl, P., et al.
Fig.4.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 5
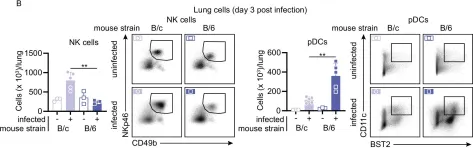
In Elife on 13 January 2022 by Gawish, R., Starkl, P., et al.
Fig.6.B

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 5
In Elife on 13 January 2022 by Gawish, R., Starkl, P., et al.
Fig.4.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 5
In Nat Commun on 3 June 2019 by Zhou, J., Huang, S., et al.
Fig.3.A

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
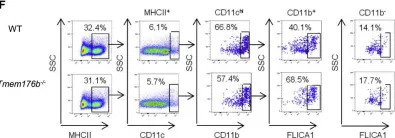
In Cancer Cell on 13 May 2019 by Segovia, M., Russo, S., et al.
Fig.2.F

-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Cancer Cell by CiteAb, provided under a CC-BY license
Image 1 of 5