Many cancers harbor pro-proliferative mutations of the mitogen-activated protein kinase (MAPK) pathway. In BRAF-driven melanoma cells treated with BRAF inhibitors, subpopulations of cells escape drug-induced quiescence through a nongenetic manner of adaptation and resume slow proliferation. Here, we found that this phenomenon is common to many cancer types driven by EGFR, KRAS, or BRAF mutations in response to multiple, clinically approved MAPK pathway inhibitors. In 2D cultures and 3D spheroid models of various cancer cell lines, a subset of cells escaped drug-induced quiescence within 4 days to resume proliferation. These "escapee" cells exhibited DNA replication deficits, accumulated DNA lesions, and mounted a stress response that depended on the ataxia telangiectasia and RAD3-related (ATR) kinase. We further identified that components of the Fanconi anemia (FA) DNA repair pathway are recruited to sites of mitotic DNA synthesis (MiDAS) in escapee cells, enabling successful completion of cell division. Analysis of patient tumor samples and clinical data correlated disease progression with an increase in DNA replication stress response factors. Our findings suggest that many MAPK pathway-mutant cancers rapidly escape drug action and that suppressing early stress tolerance pathways may achieve more durable clinical responses to MAPK pathway inhibitors.
Product Citations: 24
In Science Signaling on 1 August 2023 by Hoffman, T. E., Nangia, V., et al.
-
Biochemistry and Molecular biology
-
Genetics
In Scientific Reports on 7 October 2022 by Kim, S., Leong, A., et al.
External signaling controls cell-cycle entry until cells irreversibly commit to the cell cycle to ensure faithful DNA replication. This process is tightly regulated by cyclin-dependent kinases (CDKs) and the retinoblastoma protein (Rb). Here, using live-cell sensors for CDK4/6 and CDK2 activities, we propose that CDK4/6 initiates Rb inactivation and CDK2 activation, which coordinates the timing of cell-cycle commitment and sequential G1/S transition. Our data show that CDK4/6 activation induces Rb inactivation and thereby E2F activation, driving a gradual increase in CDK2 activity. We found that rapid CDK4/6 inhibition can reverse cell-cycle entry until CDK2 activity reaches to high levels. This suggests that high CDK2 activity is required to initiate CDK2-Rb positive feedback and CDK4/6-indpendent cell-cycle progression. Since CDK2 activation also facilitates initiation of DNA replication, the timing of CDK2-Rb positive feedback is coupled with the G1/S transition. Our experiments, which acutely increased CDK2 activity by cyclin E1 overexpression, indicate that cells commit to the cell cycle before triggering DNA replication. Together, our data suggest that CDK4/6 inactivates Rb to begin E2F and CDK2 activation, and high CDK2 activity is necessary and sufficient to generate a bistable switch for Rb phosphorylation before DNA replication. These findings highlight how cells initiate the cell cycle and subsequently commit to the cell cycle before the G1/S transition.
© 2022. The Author(s).
-
Homo sapiens (Human)
Antitumor Effects of Ral-GTPases Downregulation in Glioblastoma.
In International Journal of Molecular Sciences on 25 July 2022 by Cemeli, T., Guasch-Vallés, M., et al.
Glioblastoma (GBM) is the most common tumor in the central nervous system in adults. This neoplasia shows a high capacity of growth and spreading to the surrounding brain tissue, hindering its complete surgical resection. Therefore, the finding of new antitumor therapies for GBM treatment is a priority. We have previously described that cyclin D1-CDK4 promotes GBM dissemination through the activation of the small GTPases RalA and RalB. In this paper, we show that RalB GTPase is upregulated in primary GBM cells. We found that the downregulation of Ral GTPases, mainly RalB, prevents the proliferation of primary GBM cells and triggers a senescence-like response. Moreover, downregulation of RalA and RalB reduces the viability of GBM cells growing as tumorspheres, suggesting a possible role of these GTPases in the survival of GBM stem cells. By using mouse subcutaneous xenografts, we have corroborated the role of RalB in GBM growth in vivo. Finally, we have observed that the knockdown of RalB also inhibits cell growth in temozolomide-resistant GBM cells. Overall, our work shows that GBM cells are especially sensitive to Ral-GTPase availability. Therefore, we propose that the inactivation of Ral-GTPases may be a reliable therapeutic approach to prevent GBM progression and recurrence.
-
WB
PARP14 regulates cyclin D1 expression to promote cell-cycle progression.
In Oncogene on 1 July 2021 by O'Connor, M. J., Thakar, T., et al.
Cyclin D1 is an essential regulator of the G1-S cell-cycle transition and is overexpressed in many cancers. Expression of cyclin D1 is under tight cellular regulation that is controlled by many signaling pathways. Here we report that PARP14, a member of the poly(ADP-ribose) polymerase (PARP) family, is a regulator of cyclin D1 expression. Depletion of PARP14 leads to decreased cyclin D1 protein levels. In cells with a functional retinoblastoma (RB) protein pathway, this results in G1 cell-cycle arrest and reduced proliferation. Mechanistically, we found that PARP14 controls cyclin D1 mRNA levels. Using luciferase assays, we show that PARP14 specifically regulates cyclin D1 3'UTR mRNA stability. Finally, we also provide evidence that G1 arrest in PARP14-depleted cells is dependent on an intact p53-p21 pathway. Our work uncovers a new role for PARP14 in promoting cell-cycle progression through both cyclin D1 and the p53 pathway.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
-
Homo sapiens (Human)
-
Cancer Research
In Cell Cycle on 1 January 2021 by Bazzar, W., Bocci, M., et al.
Deregulated expression of the MYC oncogene is a frequent event during tumorigenesis and generally correlates with aggressive disease and poor prognosis. While MYC is a potent inducer of apoptosis, it often suppresses cellular senescence, which together with apoptosis is an important barrier against tumor development. For this latter function, MYC is dependent on cyclin-dependent kinase 2 (CDK2). Here, we utilized a MYC/BCL-XL-driven mouse model of acute myeloblastic leukemia (AML) to investigate whether pharmacological inhibition of CDK2 can inhibit MYC-driven tumorigenesis through induction of senescence. Purified mouse hematopoietic stem cells transduced with MYC and BCL-XL were transplanted into lethally irradiated mice, leading to the development of massive leukemia and subsequent death 15-17 days after transplantation. Upon disease onset, mice were treated with the selective CDK2 inhibitor CVT2584 or vehicle either by daily intraperitoneal injections or continuous delivery via mini-pumps. CVT2584 treatment delayed disease onset and moderately but significantly improved survival of mice. Flow cytometry revealed a significant decrease in tumor load in the spleen, liver and bone marrow of CVT2584-treated compared to vehicle-treated mice. This was correlated with induced senescence evidenced by reduced cell proliferation, increased senescence-associated β-galactosidase activity and heterochromatin foci, expression of p19ARF and p21CIP1, and reduced phosphorylation (activation) of pRb, while very few apoptotic cells were observed. In addition, phosphorylation of MYC at Ser-62 was decreased. In summary, inhibition of CDK2 delayed MYC/BCL-XL-driven AML linked to senescence induction. Our results suggest that CDK2 is a promising target for pro-senescence cancer therapy, in particular for MYC-driven tumors, including leukemia.
-
WB
-
Cancer Research
-
Cell Biology
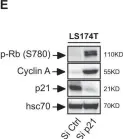
In Cell Death Dis on 27 February 2019 by Guillon, J., Petit, C., et al.
Fig.6.E

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 1