Groundwater arsenic is a notorious toxicant and exposure to environmentally relevant concentrations persists as a healthcare burden across the world. Arsenic has been reported to jeopardize the normal functioning of the immune system, but there are still gaps in the understanding of thymic T cell biology. Immunotoxic influence of arsenic in thymic integrity demands a potent restorative molecule. The objectives of this study were to examine key signaling cross-talks associated with arsenic-induced immune alterations in the thymus and propose melatonin as a potential candidate against immunological complications arising from arsenic exposure. Swiss albino mice were exposed to sodium arsenite (0.05 mg/L; in drinking water) and melatonin (IP:10 mg/kg BW) for 28 days. Melatonin successfully protected thymus from arsenic-mediated tissue degeneration and maintained immune homeostasis including T cell maturation and proliferation by mitigating oxidative stress through Nrf2 upregulation. Additionally, melatonin exerted ameliorative effect against arsenic-induced apoptosis and inflammation by inhibiting p53-mediated mitochondrial cell death pathway and NF-κB-p65/STAT3-mediated proinflammatory pathway, respectively. For the first time, we showed that arsenic-induced profibrotic changes were inhibited by melatonin through targeting of inflammation-associated EMT. Our findings clearly demonstrate that melatonin can be a viable and promising candidate in combating arsenic-induced immune toxicity with no collateral damage, making it an important research target.
© 2024 International Union of Biochemistry and Molecular Biology.
Product Citations: 12
In BioFactors (Oxford, England) on 4 August 2024 by Das, A., Mitra, A., et al.
-
Immunology and Microbiology
Packed red blood cells inhibit T-cell activation via ROS-dependent signaling pathways.
In The Journal of Biological Chemistry on 8 March 2021 by Gerner, M. C., Bileck, A., et al.
Numerous observations indicate that red blood cells (RBCs) affect T-cell activation and proliferation. We have studied effects of packed RBCs (PRBCs) on T-cell receptor (TCR) signaling and the molecular mechanisms whereby (P)RBCs modulate T-cell activation. In line with previous reports, PRBCs attenuated the expression of T-cell activation markers CD25 and CD69 upon costimulation via CD3/CD28. In addition, T-cell proliferation and cytokine expression were markedly reduced when T-cells were stimulated in the presence of PRBCs. Inhibitory activity of PRBCs required direct cell-cell contact and intact PRBCs. The production of activation-induced cellular reactive oxygen species, which act as second messengers in T-cells, was completely abrogated to levels of unstimulated T-cells in the presence of PRBCs. Phosphorylation of the TCR-related zeta chain and thus proximal TCR signal transduction was unaffected by PRBCs, ruling out mechanisms based on secreted factors and steric interaction restrictions. In large part, downstream signaling events requiring reactive oxygen species for full functionality were affected, as confirmed by an untargeted MS-based phosphoproteomics approach. PRBCs inhibited T-cell activation more efficiently than treatment with 1 mM of the antioxidant N-acetyl cysteine. Taken together, our data imply that inflammation-related radical reactions are modulated by PRBCs. These immunomodulating effects may be responsible for clinical observations associated with transfusion of PRBCs.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
-
Biochemistry and Molecular biology
-
Cardiovascular biology
-
Immunology and Microbiology
In Journal of the American Society of Nephrology : JASN on 1 November 2020 by Brandt, S., Ballhause, T. M., et al.
Kidney injuries that result in chronic inflammation initiate crosstalk between stressed resident cells and infiltrating immune cells. In animal models, whole-body receptor Notch3 deficiency protects from leukocyte infiltration and organ fibrosis. However, the relative contribution of Notch3 expression in tissue versus infiltrating immune cells is unknown.
Chimeric mice deficient for Notch3 in hematopoietic cells and/or resident tissue cells were generated, and kidney fibrosis and inflammation after unilateral ureteral obstruction (UUO) were analyzed. Adoptive transfer of labeled bone marrow-derived cells validated the results in a murine Leishmania ear infection model. In vitro adhesion assays, integrin activation, and extracellular matrix production were analyzed.
Fibrosis follows UUO, but inflammatory cell infiltration mostly depends upon Notch3 expression in hematopoietic cells, which coincides with an enhanced proinflammatory milieu (e.g., CCL2 and CCL5 upregulation). Notch3 expression on CD45+ leukocytes plays a prominent role in efficient cell transmigration. Functionally, leukocyte adhesion and integrin activation are abrogated in the absence of receptor Notch3. Chimeric animal models also reveal that tubulointerstitial fibrosis develops, even in the absence of prominent leukocyte infiltrates after ureteral obstruction. Deleting Notch3 receptors on resident cells blunts kidney fibrosis, ablates NF-κB signaling, and lessens matrix deposition.
Cell-specific receptor Notch3 signaling independently orchestrates leukocyte infiltration and organ fibrosis. Interference with Notch3 signaling may present a novel therapeutic approach in inflammatory as well as fibrotic diseases.
Copyright © 2020 by the American Society of Nephrology.
-
Endocrinology and Physiology
-
Immunology and Microbiology
In Molecular and Cellular Biology on 15 November 2018 by Wabnitz, G. H., Kirchgessner, H., et al.
While several protein serine/threonine kinases control cytokine production by T cells, the roles of serine/threonine phosphatases are largely unexplored. Here, we analyzed the involvement of protein phosphatase 1α (PP1α) in cytokine synthesis following costimulation of primary human T cells. Small interfering RNA (siRNA)-mediated knockdown of PP1α (PP1KD) or expression of a dominant negative PP1α (D95N-PP1) drastically diminished interleukin-10 (IL-10) production. Focusing on a key transcriptional activator of human IL-10, we demonstrate that nuclear translocation of NF-κB was significantly inhibited in PP1KD or D95N-PP1 cells. Interestingly, knockdown of cofilin, a known substrate of PP1 containing a nuclear localization signal, also prevented nuclear accumulation of NF-κB. Expression of a constitutively active nonphosphorylatable S3A-cofilin in D95N-PP1 cells restored nuclear translocation of NF-κB and IL-10 expression. Subpopulation analysis revealed that defective nuclear translocation of NF-κB was most prominent in CD4+ CD45RA- CXCR3- T cells that included IL-10-producing TH2 cells. Together these findings reveal novel functions for PP1α and its substrate cofilin in T cells namely the regulation of the nuclear translocation of NF-κB and promotion of IL-10 production. These data suggest that stimulation of PP1α could limit the overwhelming immune responses seen in chronic inflammatory diseases.
Copyright © 2018 American Society for Microbiology.
-
Cell Biology
-
Immunology and Microbiology
In Cell Stem Cell on 3 August 2017 by Takizawa, H., Fritsch, K., et al.
Bacterial infection leads to consumption of short-lived innate immune effector cells, which then need to be replenished from hematopoietic stem and progenitor cells (HSPCs). HSPCs express pattern recognition receptors, such as Toll-like receptors (TLRs), and ligation of these receptors induces HSPC mobilization, cytokine production, and myeloid differentiation. The underlying mechanisms involved in pathogen signal transduction in HSCs and the resulting biological consequences remain poorly defined. Here, we show that in vivo lipopolysaccharide (LPS) application induces proliferation of dormant HSCs directly via TLR4 and that sustained LPS exposure impairs HSC self-renewal and competitive repopulation activity. This process is mediated via TLR4-TRIF-ROS-p38, but not MyD88 signaling, and can be inhibited pharmacologically without preventing emergency granulopoiesis. Live Salmonella Typhimurium infection similarly induces proliferative stress in HSCs, in part via TLR4-TRIF signals. Thus, while direct TLR4 activation in HSCs might be beneficial for controlling systemic infection, prolonged TLR4 signaling has detrimental effects and may contribute to inflammation-associated HSPC dysfunction.
Copyright © 2017 Elsevier Inc. All rights reserved.
-
Immunology and Microbiology
-
Stem Cells and Developmental Biology
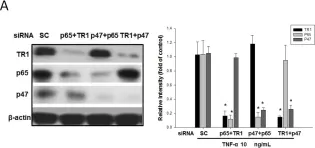
In PLoS One on 2 December 2015 by Lin, C. P., Huang, P. H., et al.
Fig.7.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 1