The cycling intestinal stem cells (ISCs) exhibit radiosensitivity, and their death or impaired regenerative capacity following irradiation may result in intestinal barrier dysfunction. The cystathionine-β-synthase (CBS)/H2S axis plays a critical role in regulating cell proliferation, reactive oxygen species scavenging, and the DNA damage response. However, it remains unclear whether the CBS/H2S axis modulates ISC homeostasis and tissue radiosensitivity.
Intestinal epithelium specific conditional CBS knockout mice were generated by crossing CBSfl/+ mice with Villin-CreERT2 mice. CAGGCre-ER™ mice were crossed with CBSfl/fl mice to achieve CBS knockout in multiple tissues and cell types. The Lgr5-Tdtaomato-Flag mice were generated by CRISPR/Cas9 system. The CBS inhibitor AOAA or the H2S donor GYY4137 was used to treat mice or intestinal crypt organoids. Hematoxylin and eosin, immunohistochemistry, immunofluorescence, Western blot, qRT-PCR, et al. were employed to investigate the role of the CBS/H2S axis in ISCs homeostasis and radiation-induced intestinal damage.
Lgr5 + ISCs and progenitor cells expressed higher levels of CBS than differentiated cells. The cecum and colon expressed significant higher CBS levels than the small intestine. Treatment with the H2S donor GYY4137 enhanced the proliferation of intestinal organoids in vitro, while inhibition of CBS by AOAA reduced this effect. Genetic knockout of CBS in the intestinal epithelium or global downregulation of CBS driven by CAGG-CreER™ in vivo did not affect ISC proliferation or differentiation under physiological conditions. Pharmacological regulation of the CBS/H2S axis in vitro failed to protect organoids from radiation-induced damage. Interestingly, administration of AOAA in vivo reduced radiation-induced atrophy of the intestinal mucosa. Furthermore, global downregulation of CBS significantly promoted ISC recovery after irradiation exposure. However, intestinal epithelium-specific CBS knockout did not confer radioprotective effects.
Our findings suggest that the CBS/H2S axis contributes to the regulation of ISC homeostasis and represents a potential target for radiation protection, mediated through the intervention of non-epithelial cells.
© 2025. The Author(s).
Product Citations: 28
The CBS/H2S axis regulates intestinal stem cell homeostasis and radiation-induced intestinal damage.
In Stem Cell Research & Therapy on 31 October 2025 by Wu, T., Zheng, Z., et al.
-
Stem Cells and Developmental Biology
Maternal stress triggers early-life eczema through fetal mast cell programming.
In Nature on 1 October 2025 by Serhan, N., Abdullah, N. S., et al.
Prenatal stress (PS) is a repeated exposure to aversive situations during pregnancy, including high emotional strain, which is suspected to affect homeostatic systems in infants. Paediatric eczema develops quickly after birth at flexural sites subjected to continuous mechanical constraints1,2. Although epidemiological studies have suggested an association between PS and a higher risk of eczema in children3-6, no causative biological link has yet been identified. Here we show that eczema at birth originates from molecular dysregulations of neuroimmune circuits in utero, triggered by fluctuations in the maternal hypothalamic-pituitary-adrenal axis. We found that offspring of stressed pregnant dams have dysregulated mast cells and skin-projecting neurons and quickly develop eczema in response to harmless mechanical friction. We demonstrated that PS transiently modulates amniotic fluid corticosterone concentrations, which directly alters the activation program of skin mast cells expressing the glucocorticoid receptor Nr3c1 and the adjacent sensory neurons conveying mechanosensation. Therapeutic normalization of maternal corticosterone concentrations or genetic depletion of Mcpt5+ mast cells during stressed gestation prevents fetal immune dysregulation and protects against eczema development after birth. Our findings support a new model in which early-onset paediatric eczema originates from dysregulations in the fetal immune system, caused by fluctuations in maternal glucocorticoids induced by stress.
© 2025. The Author(s).
-
FC/FACS
In IScience on 18 April 2025 by Pioli, K. T., Ghosh, S., et al.
Mutations that negatively impact mitochondrial function are highly prevalent in humans and lead to disorders with a wide spectrum of disease phenotypes, including deficiencies in immune cell development and/or function. Previous analyses of mice with a hepatocyte-specific cytochrome c oxidase (COX) deficiency revealed an unexpected peripheral blood leukopenia associated with splenic and thymic atrophy. Here, we use mice with a hepatocyte-specific deletion of the COX assembly factor Sco1 to show that metabolic defects extrinsic to the hematopoietic compartment lead to a pan-lymphopenia represented by severe losses in both B and T cells. We further demonstrate that immune defects in these mice are associated with the loss of bone marrow lymphoid progenitors common to both lineages and early signs of autoantibody-mediated autoimmunity. Our findings collectively identify hepatocyte dysfunction as a potential instigator of immunodeficiency in patients with congenital mitochondrial defects who suffer from chronic or recurrent infections.
© 2025 The Author(s).
In Cell Reports on 25 March 2025 by Ravi, V. R., Korkmaz, F. T., et al.
Neutrophilic asthma is a vexing disease, but mechanistic and therapeutic advancements will require better models of allergy-induced airway neutrophilia. Here, we find that periodic ovalbumin (OVA) inhalation in sensitized mice elicits rapid allergic airway inflammation and pathophysiology mimicking neutrophilic asthma. OVA-experienced murine lungs harbor diverse clusters of CD4+ resident memory T (TRM) cells, including unconventional RORγtnegative/low T helper 17 (TH17) cells. Acute OVA challenge instigates interleukin (IL)-17A secretion from these TRM cells, driving CXCL5 production from Muc5achigh airway secretory cells, leading to destructive airway neutrophilia. The TRM and epithelial cell signals discovered herein are also observed in adult human asthmatic airways. Epithelial antigen presentation regulates this biology by skewing TRM cells toward TH2 and TH1 fates so that TH1-related interferon (IFN)-γ suppresses IL-17A-driven, CXCL5-mediated airway neutrophilia. Concordantly, in vivo IFN-γ supplementation improves disease outcomes. Thus, using our model of neutrophilic asthma, we identify lung epithelial-CD4+ TRM cell crosstalk as a key rheostat of allergic airway neutrophilia.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
-
Immunology and Microbiology
In Cell Reports Medicine on 19 November 2024 by Li, S., Tao, B., et al.
Myocardial infarction (MI) results in aberrant cardiac metabolism, but no therapeutics have been designed to target cardiac metabolism to enhance heart repair. We engineer a humanized monoclonal antibody against the ectonucleotidase ENPP1 (hENPP1mAb) that targets metabolic crosstalk in the infarcted heart. In mice expressing human ENPP1, systemic administration of hENPP1mAb metabolically reprograms myocytes and non-myocytes and leads to a significant rescue of post-MI heart dysfunction. Using metabolomics, single-nuclear transcriptomics, and cellular respiration studies, we show that the administration of the hENPP1mAb induces organ-wide metabolic and transcriptional reprogramming of the heart that enhances myocyte cellular respiration and decreases cell death and fibrosis in the infarcted heart. Biodistribution and safety studies showed specific organ-wide distribution with the antibody being well tolerated. In humanized animals, with drug clearance kinetics similar to humans, we demonstrate that a single "shot" of the hENPP1mAb after MI is sufficient to rescue cardiac dysfunction.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Cardiovascular biology
-
Cell Biology
In Elife on 17 April 2018 by Kasler, H. G., Lee, I. S., et al.
Fig.1.E
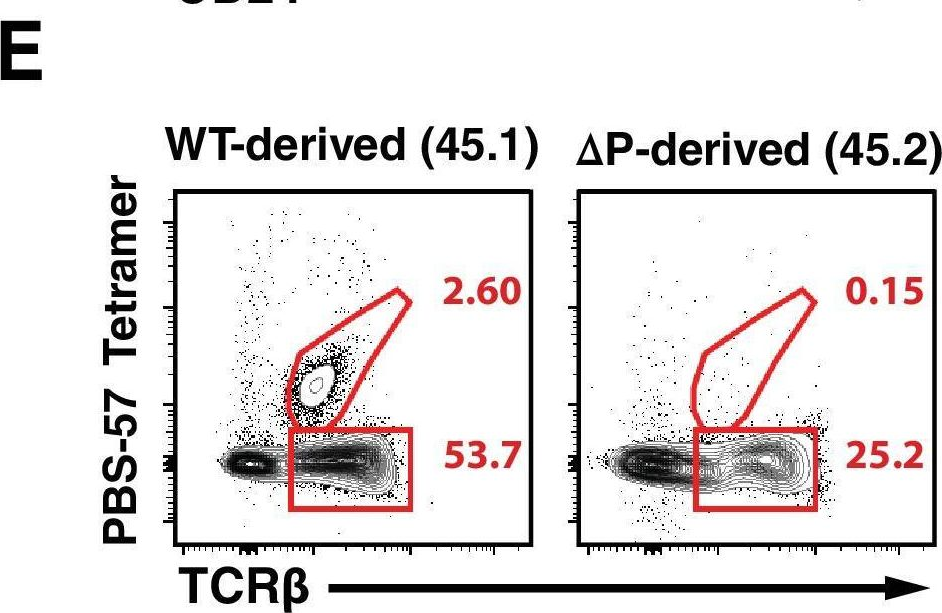
-
FC/FACS
-
Collected and cropped from eLife by CiteAb, provided under a CC-BY license
Image 1 of 2
In Elife on 17 April 2018 by Kasler, H. G., Lee, I. S., et al.
Fig.3.D
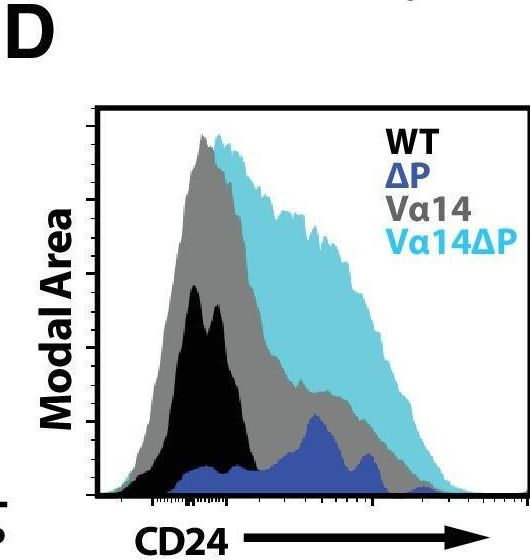
-
FC/FACS
-
Collected and cropped from eLife by CiteAb, provided under a CC-BY license
Image 1 of 2