Resistance to chemotherapy remains a major hurdle to the cure of patients with acute myeloid leukemia (AML). Recent studies indicate that a minority of malignant cells, termed drug-tolerant persisters (DTP), stochastically upregulate stress pathways to evade cell death upon acute exposure to chemotherapy without acquiring new genetic mutations. This chemoresistant state is transient and the cells return to the baseline state after removal of chemotherapy. Nevertheless, the mechanisms employed by DTP to resist chemotherapy are not well understood and it is largely unknown whether these mechanisms are also seen in patients receiving chemotherapy. Here, we used leukemia cell lines, primary AML patients' samples and samples from patients with AML receiving systemic chemotherapy to study the DTP state. We demonstrated that a subset of AML cells transiently increases membrane rigidity to resist killing due to acute exposure to daunorubicin and Ara-C. Upon removal of the chemotherapy, membrane rigidity returned to baseline and the cells regained chemosensitivity. Although resistant to chemotherapy, the increased membrane rigidity rendered AML cells more susceptible to T-cell-mediated killing. Thus, we identified a novel mechanism by which DTP leukemic cells evade chemotherapy and a strategy to eradicate these persistent cells.
Product Citations: 52
In Haematologica on 1 April 2025 by Morgenstern, Y., Lee, J., et al.
-
Homo sapiens (Human)
-
Cancer Research
-
Cardiovascular biology
-
Immunology and Microbiology
In Aging Cell on 1 March 2025 by Vandiver, A. R., Torres, A., et al.
The mitochondrial genome (mtDNA) is an important source of inherited extranuclear variation. Clonal increases in mtDNA mutation heteroplasmy have been implicated in aging and disease, although the impact of this shift on cell function is challenging to assess. Reprogramming to pluripotency affects mtDNA mutation heteroplasmy. We reprogrammed three human fibroblast lines with known heteroplasmy for deleterious mtDNA point or deletion mutations. Quantification of mutation heteroplasmy in the resulting 76 induced pluripotent stem cell (iPSC) clones yielded a bimodal distribution, creating three sets of clones with high levels or absent mutation heteroplasmy with matched nuclear genomes. iPSC clones with elevated deletion mutation heteroplasmy show altered growth dynamics, which persist in iPSC-derived progenitor cells. We identify transcriptomic and metabolic shifts consistent with increased investment in neutral lipid synthesis as well as increased epigenetic age in high mtDNA deletion mutation iPSC, consistent with changes occurring in cellular aging. Together, these data demonstrate that high mtDNA mutation heteroplasmy induces changes occurring in cellular aging.
© 2024 The Author(s). Aging Cell published by Anatomical Society and John Wiley & Sons Ltd. This article has been contributed to by U.S. Government employees and their work is in the public domain in the USA.
-
FC/FACS
-
Homo sapiens (Human)
-
Cell Biology
-
Stem Cells and Developmental Biology
BMSC-derived exosomes promote osteoporosis alleviation via M2 macrophage polarization.
In Molecular Medicine on 19 November 2024 by Zhang, Y., Bai, J., et al.
Osteoporosis is characterized by reduced bone mass due to imbalanced bone metabolism. Exosomes derived from bone mesenchymal stem cells (BMSCs) have been shown to play roles in various diseases. This study aimed to clarify the regulatory function and molecular mechanism of BMSCs-derived exosomes in osteogenic differentiation and their potential therapeutic effects on osteoporosis. Exosomes were extracted from BMSCs. Bone marrow-derived macrophages (BMDMs) were cultured and internalized with BMSCs-derived exosomes. Real-time quantitative PCR was used to detect the expression of macrophage surface markers and tripartite motif (TRIM) family genes. BMDMs were co-cultured with human osteoblasts to assess osteogenic differentiation. Western blot was performed to analyze the ubiquitination of triggering receptor expressed on myeloid cell 1 (TREM1) mediated by TRIM25. An ovariectomized mice model was established to evaluate the role of TRIM25 and exosomes in osteoporosis. Exosomes were successfully isolated from BMSCs. BMSCs-derived exosomes upregulated TRIM25 expression, promoting M2 macrophage polarization and osteogenic differentiation. TRIM25 facilitated the ubiquitination and degradation of TREM1. Overexpression of TREM1 reversed the enhanced M2 macrophage polarization and osteogenic differentiation caused by TRIM25 overexpression. TRIM25 enhanced the protective effect of BMSCs-derived exosomes against bone loss in mice. These findings suggested that BMSCs-derived exosomes promoted osteogenic differentiation by regulating M2 macrophage polarization through TRIM25-mediated ubiquitination and degradation of TREM1. This mechanism might provide a novel approach for treating osteoporosis.
© 2024. The Author(s).
-
FC/FACS
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Immunology and Microbiology
In The Journal of Biological Chemistry on 1 August 2024 by Ramani, M., Singh, R. K., et al.
Lenalidomide, a thalidomide derivative, is prescribed as maintenance therapy for multiple myeloma (MM). Patients with MM receiving lenalidomide were found to develop a distinct therapy-related B cell acute lymphoblastic leukemia (B-ALL). However, the molecular mechanism by which lenalidomide drives B-ALL is unknown. We show that thalidomide treatment of B cell lines increased CD34 expression and fibronectin adhesion. This resembled the effects of Ikzf1 loss of function mutations in B-ALL. IKZF1 is a transcription factor that can act as both a transcriptional activator and a repressor depending upon the target loci. In our experiments, thalidomide-induced degradation of IKZF1 increased the expression of its transcriptional repression targets Itga5 and CD34 explaining the increased adhesion and stemness. Strikingly, withdrawal of thalidomide lead to the mis-localization of IKZF1 to the cytoplasm. Moreover, chromatin immunoprecipitation data showed a long-term effect of thalidomide treatment on IKZF1 target loci. This included decreased chromatin occupancy at early B cell factor 1 (EBF1) and Spi1 (PU.1). Consequently, B-cell lineage specifying transcription factors including Pax5, Spi1 and EBF1 were downregulated even after 7 days of thalidomide withdrawal. Our study thus provides a molecular mechanism of thalidomide-induced B-ALL whereby thalidomide alters the chromatin occupancy of IKZF1 at key B-cell lineage transcription factors leading to a persistent block in B-cell differentiation.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
-
FC/FACS
-
Biochemistry and Molecular biology
-
Cancer Research
-
Immunology and Microbiology
CD37 is a safe chimeric antigen receptor target to treat acute myeloid leukemia.
In Cell Reports Medicine on 18 June 2024 by Caulier, B., Joaquina, S., et al.
Acute myeloid leukemia (AML) is characterized by the accumulation of immature myeloid cells in the bone marrow and the peripheral blood. Nearly half of the AML patients relapse after standard induction therapy, and new forms of therapy are urgently needed. Chimeric antigen receptor (CAR) T therapy has so far not been successful in AML due to lack of efficacy and safety. Indeed, the most attractive antigen targets are stem cell markers such as CD33 or CD123. We demonstrate that CD37, a mature B cell marker, is expressed in AML samples, and its presence correlates with the European LeukemiaNet (ELN) 2017 risk stratification. We repurpose the anti-lymphoma CD37CAR for the treatment of AML and show that CD37CAR T cells specifically kill AML cells, secrete proinflammatory cytokines, and control cancer progression in vivo. Importantly, CD37CAR T cells display no toxicity toward hematopoietic stem cells. Thus, CD37 is a promising and safe CAR T cell AML target.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
-
FC/FACS
-
Homo sapiens (Human)
-
Cancer Research
-
Immunology and Microbiology
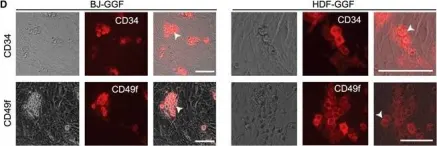
In Cell Rep on 4 December 2018 by Gomes, A. M., Kurochkin, I., et al.
Fig.1.D

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from Cell Rep by CiteAb, provided under a CC-BY license
Image 1 of 2
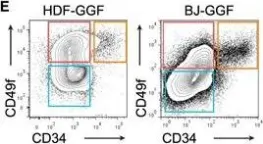
In Cell Rep on 4 December 2018 by Gomes, A. M., Kurochkin, I., et al.
Fig.1.E

-
FC/FACS
-
Homo sapiens (Human)
Collected and cropped from Cell Rep by CiteAb, provided under a CC-BY license
Image 1 of 2