
Acute kidney injury (AKI) is a life-threating syndrome characterized by sudden loss of kidney function, and its management is challenging and often suboptimal. Mesenchymal stem cells (MSCs) have shown promise in AKI therapy in pre-clinical and clinical trials; however, their clinical application still faces many challenges. MSC-derived small extracellular vesicles (sEV) may help overcome these challenges. In the current study, we overexpressed Klotho in MSCs and then isolated Klotho-loaded sEV (Klotho-sEV) using anion-exchange chromatography. Klotho-sEV displayed characteristics comparable to those of sEV in terms of size, morphology, conventional markers, and biosafety, as well as a higher abundance of Klotho protein. In rhabdomyolysis-induced AKI, sEV showed preferential tropism in injured kidneys. We found significantly and stably accelerated renal recovery, mitigated functional and histological abnormalities, stimulated tubular cell proliferation, reduced injury and inflammatory marker expression, and restored endogenous Klotho loss in mice after the administration of Klotho-sEV. In addition, Klotho-sEV treatment activated the mTOR and MEK1/2 signaling pathways. Proteomics and small RNA sequencing analyses of sEV and Klotho-sEV revealed abundant proteins and miRNAs involved in anti-inflammation and reno-protection, and Klotho-sEV showed characteristics that were different from those of sEV. In conclusion, Klotho-sEV may be a promising cell-free strategy for the treatment of AKI.
© 2025. The Author(s).
Product Citations: 26
In Journal of Nanobiotechnology on 7 June 2025 by Deng, X. H., Wu, Z. C., et al.
-
Stem Cells and Developmental Biology
Evaluated NSUN3 in reticulocytes from HbH-CS disease that reflects cellular stress in erythroblasts.
In Annals of Hematology on 1 April 2025 by Liu, H., Peng, C., et al.
Hemoglobin H Disease-Constant Spring (HbH-CS) represents a severe variant of α-thalassemia characterized by a fundamental pathological mechanism involving inadequate synthesis of α-globin chains. This deficiency results in the formation of unstable Hemoglobin H (HbH) due to the aggregation of free β-globin chains, which subsequently induces an imbalance in oxidative stress within erythrocytes. This imbalance leads to an abnormal accumulation of reactive oxygen species (ROS), which in turn promotes lipid peroxidation, culminating in the production of malondialdehyde (MDA) and a significant depletion of glutathione (GSH). Concurrently, Nrf2 is translocated to the nucleus, where it activates the antioxidant response element (ARE) to mitigate cellular stress. Here, we report that NSUN3 (which, together with ALKBH1, maintains mitochondrial function through m5C→f5C modification) is abnormally overexpressed in reticulocytes from patients with HbH-CS, and an in vitro cellular model of NSUN3 overexpression/silencing was constructed using K562 cells, which have the potential for erythroid lineage differentiation and retain an intact cluster of bead protein genes. Functional assays indicated that the overexpression of NSUN3 significantly intensified the accumulation of intracellular ROS and MDA, led to a reduction in GSH levels, and diminished the overall cellular antioxidant capacity (T-AOC). This may be due to ROS accumulation resulting from inhibition of mitochondrial respiratory chain complex I, II, and IV synthesis through aberrant m5C→f5C modification. In addition, NSUN3 overexpression further exacerbates oxidative stress by inhibiting the phosphorylation of Nrf2 hindering its translocation into the nucleus and weakening the cellular antioxidant system. Moreover, we also observed that NSUN3 overexpression exacerbated intracellular DNA damage and inhibited cellular value-added activity, and silencing NSUN3 showed the opposite result. Our research offers initial insights into the molecular mechanisms through which NSUN3 modulates oxidative stress in erythrocytes via its role in epigenetic modifications. These findings contribute to a deeper understanding of the clinical management of patients with Hb H-CS.
© 2025. The Author(s).
In Clinical Cancer Research on 1 November 2024 by Freeland, J., Muñoz, M., et al.
High-grade complex karyotype sarcomas are a heterogeneous group of tumors with a uniformly poor prognosis. Within complex karyotype sarcomas, there are innumerable genetic changes but identifying those that are clinically relevant has been challenging.
To address this, we utilized a pooled genetic screening approach, informed by The Cancer Genome Atlas (TCGA) data, to identify key drivers and modifiers of sarcoma development that were validated in vivo.
YAP1 and wild-type KRAS were validated as drivers and transformed human mesenchymal stem cells into two distinct sarcoma subtypes, undifferentiated pleomorphic sarcoma and myxofibrosarcoma, respectively. A subset of tumors driven by CDK4 and PIK3CA reflected leiomyosarcoma and osteosarcoma demonstrating the plasticity of this approach and the potential to investigate sarcoma subtype heterogeneity. All generated tumors histologically reflected human sarcomas and had increased aneuploidy as compared to simple karyotype sarcomas. Comparing differential gene expression of TCGA samples to model data identified increased oxidative phosphorylation signaling in YAP1 tumors. Treatment of a panel of soft tissue sarcomas with a combination of YAP1 and oxidative phosphorylation inhibitors led to significantly decreased viability.
Transcriptional co-analysis of TCGA patient samples to YAP1 and KRAS model tumors supports that these sarcoma subtypes lie along a spectrum of disease and adds guidance for further transcriptome-based refinement of sarcoma subtyping. This approach can be used to begin to understand pathways and mechanisms driving human sarcoma development, the relationship between sarcoma subtypes, and to identify and validate new therapeutic vulnerabilities for this aggressive and heterogeneous disease.
©2024 American Association for Cancer Research.
-
Cancer Research
-
Genetics
In Bioactive Materials on 1 April 2024 by Ouyang, Y., Hong, Y., et al.
Abdominal Aortic Aneurysm (AAA) is a life-threatening vascular disease characterized by the weakening and ballooning of the abdominal aorta, which has no effective therapeutic approaches due to unclear molecular mechanisms. Using single-cell RNA sequencing, we analyzed the molecular profile of individual cells within control and AAA abdominal aortas. We found cellular heterogeneity, with increased plasmacytoid dendritic cells and reduced endothelial cells and vascular smooth muscle cells (VSMCs) in AAA. Up-regulated genes in AAA were associated with muscle tissue development and apoptosis. Genes controlling VSMCs aberrant switch from contractile to synthetic phenotype were significantly enriched in AAA. Additionally, VSMCs in AAA exhibited cell senescence and impaired oxidative phosphorylation. Similar observations were made in a mouse model of AAA induced by Angiotensin II, further affirming the relevance of our findings to human AAA. The concurrence of gene expression changes between human and mouse highlighted the impairment of oxidative phosphorylation as a potential target for intervention. Nicotinamide phosphoribosyltransferase (NAMPT, also named VISFATIN) signaling emerged as a signature event in AAA. NAMPT was significantly downregulated in AAA. NAMPT-extracellular vesicles (EVs) derived from mesenchymal stem cells restored NAMPT levels, and offered protection against AAA. Furthermore, NAMPT-EVs not only repressed injuries, such as cell senescence and DNA damage, but also rescued impairments of oxidative phosphorylation in both mouse and human AAA models, suggesting NAMPT supplementation as a potential therapeutic approach for AAA treatment. These findings shed light on the cellular heterogeneity and injuries in AAA, and offered promising therapeutic intervention for AAA treatment.
© 2023 The Authors.
-
FC/FACS
In Nature Biomedical Engineering on 1 April 2024 by Harding, J., Vintersten-Nagy, K., et al.
The immunogenicity of transplanted allogeneic cells and tissues is a major hurdle to the advancement of cell therapies. Here we show that the overexpression of eight immunomodulatory transgenes (Pdl1, Cd200, Cd47, H2-M3, Fasl, Serpinb9, Ccl21 and Mfge8) in mouse embryonic stem cells (mESCs) is sufficient to immunologically 'cloak' the cells as well as tissues derived from them, allowing their survival for months in outbred and allogeneic inbred recipients. Overexpression of the human orthologues of these genes in human ESCs abolished the activation of allogeneic human peripheral blood mononuclear cells and their inflammatory responses. Moreover, by using the previously reported FailSafe transgene system, which transcriptionally links a gene essential for cell division with an inducible and cell-proliferation-dependent kill switch, we generated cloaked tissues from mESCs that served as immune-privileged subcutaneous sites that protected uncloaked allogeneic and xenogeneic cells from rejection in immune-competent hosts. The combination of cloaking and FailSafe technologies may allow for the generation of safe and allogeneically accepted cell lines and off-the-shelf cell products.
© 2023. The Author(s).
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
-
Stem Cells and Developmental Biology
In Stem Cell Res Ther on 22 January 2019 by Gao, K., Kumar, P., et al.
Fig.1.D

-
FC/FACS
-
Homo sapiens (Human)
Collected and cropped from Stem Cell Res Ther by CiteAb, provided under a CC-BY license
Image 1 of 2
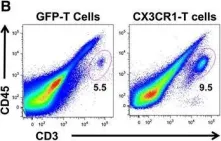
In J Immunother Cancer on 21 April 2016 by Siddiqui, I., Erreni, M., et al.
Fig.3.B

-
FC/FACS
-
Homo sapiens (Human)
Collected and cropped from J Immunother Cancer by CiteAb, provided under a CC-BY license
Image 1 of 2