Phosphatase and tensin homolog (PTEN) is vital for B cell development, acting as a key negative regulator in the PI3K signaling pathway. We used CD23-cre to generate PTEN-conditional knockout mice (CD23-cKO) to examine the impact of PTEN mutation on peripheral B cells. Unlike mb1-cre-mediated PTEN deletion in early B cells, CD23-cKO mutants exhibited systemic inflammation with increased IL-6 production in mature B cells upon CpG stimulation. Inflammatory B cells in CD23-cKO mice showed elevated phosphatidylinositol 3-phosphate [PI(3)P] levels and increased TLR9 endosomal localization. Pharmacological inhibition of PI(3)P synthesis markedly reduced TLR9-mediated IL-6. Single-cell RNA-sequencing (RNA-seq) revealed altered endocytosis, BANK1, and NF-κB1 expression in PTEN-deficient B cells. Ectopic B cell receptor (BCR) expression on non-inflammatory mb1-cKO B cells restored BANK1 and NF-κB1 expression, enhancing TLR9-mediated IL-6 production. Our study highlights PTEN as a crucial inflammatory checkpoint, regulating TLR9/IL-6 axis by fine-tuning PI(3)P homeostasis. Additionally, BCR downregulation prevents the differentiation of inflammatory B cells in PTEN deficiency.
© 2024 The Author(s).
Product Citations: 7
PTEN acts as a crucial inflammatory checkpoint controlling TLR9/IL-6 axis in B cells.
In IScience on 19 July 2024 by Tsai, P. J., Chen, M. Y., et al.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
Disparate macrophage responses are linked to infection outcome of Hantan virus in humans or rodents.
In Nature Communications on 10 January 2024 by Ma, H., Yang, Y., et al.
Hantaan virus (HTNV) is asymptomatically carried by rodents, yet causes lethal hemorrhagic fever with renal syndrome in humans, the underlying mechanisms of which remain to be elucidated. Here, we show that differential macrophage responses may determine disparate infection outcomes. In mice, late-phase inactivation of inflammatory macrophage prevents cytokine storm syndrome that usually occurs in HTNV-infected patients. This is attained by elaborate crosstalk between Notch and NF-κB pathways. Mechanistically, Notch receptors activated by HTNV enhance NF-κB signaling by recruiting IKKβ and p65, promoting inflammatory macrophage polarization in both species. However, in mice rather than humans, Notch-mediated inflammation is timely restrained by a series of murine-specific long noncoding RNAs transcribed by the Notch pathway in a negative feedback manner. Among them, the lnc-ip65 detaches p65 from the Notch receptor and inhibits p65 phosphorylation, rewiring macrophages from the pro-inflammation to the pro-resolution phenotype. Genetic ablation of lnc-ip65 leads to destructive HTNV infection in mice. Thus, our findings reveal an immune-braking function of murine noncoding RNAs, offering a special therapeutic strategy for HTNV infection.
© 2024. The Author(s).
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
Preprint on Research Square on 14 March 2023 by Tsai, P., Hsu, W., et al.
Phosphatase and tensin homolog (PTEN) is a negative regulator for PI3K signaling essential for B cell development. To explore the physiological effects of PTEN mutation on peripheral B cells, we generated CD23/cre-PTEN Flox/Flox (CD23-cKO) mice in this study to avoid the developmental arrest. The mutant mice develop systemic inflammation associated with B cell expansion in the early phase followed with a severe immune cell-infiltration in multiple vital organs. PTEN deficiency leads to an accumulation of PI(3)P, an increase of lysosomal recruitment of TLR9/p38 complex, and an aberrant activation of TLR9/IL-6 axis in B cells. Interestingly, cholesterol biosynthesis pathway is upregulated in mutant cells upon TLR9 engagement. A blockade of cholesterol biosynthesis by targeting SQLE greatly reduces the level of PI(3)P and the interaction between TLR9 and p38, which lowers the level of TLR9-induced IL-6. Thus, PTEN represents a critical metabolic checkpoint that fine-tunes lipid and cholesterol homeostasis to control TLR9-driven inflammation.
-
FC/FACS
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Cell Biology
-
Immunology and Microbiology
Preprint on Research Square on 6 January 2022 by Zhang, F., Ma, H., et al.
Hantaan virus (HTNV) is principally maintained and transmitted by rodents in nature, the infection of which is non-pathogenic in the field or laboratory mouse, but can cause hemorrhagic fever with renal syndrome (HFRS) in human beings, a severe systemic inflammatory disease with high mortality. It remains obscure how HTNV infection leads to disparate outcomes in distinct species. Here, we revealed a differential immune status in murine versus humans post HTNV infection, which was orchestrated by the macrophage reprogramming process and characterized by late-phase inactivation of NF-κB signaling. In HFRS patients, the immoderate and continuous activation of inflammatory monocyte/macrophage (M1) launched TNFα-centered cytokine storm and aggravated host immunopathologic injury, which can be life-threatening; however, in field or laboratory mice, the M1 activation and TNFα release were significantly suppressed at the late infection stage of HTNV, restricting excessive inflammation and blocking viral disease process, which also protected mice from secondary LPS challenge or polymicrobial sepsis. Mechanistically, we found that murine macrophage phenotype was dynamically manipulated by HTNV via the Notch-lncRNA-p65 axis. At the early stage of HTNV infection, the intracellular domain of Notch receptor (NICD) was activated by viral nucleocapsid (NP) stimulation and potentiated the NF-κB pathway by associating with and facilitating the interaction between IKKβ and p65. At the late stage, Notch signaling launched the expression of diverse murine-specific long non-coding RNAs (lncRNAs) and attenuated M1 polarization. Among them, lncRNA 30740.1 (termed as lnc-ip65, an inhibitor of p65) bound to p65 and hindered its phosphorylation, exerting negative feedback on the NF-κB pathway. Genetic ablation of lnc-ip65 shifted the balance of macrophage polarization from a pro-resolution to an inflammatory phenotype, leading to superabundant production of pro-inflammatory cytokines and increasing mice susceptibility to HTNV infection or bacterial sepsis. Collectively, our findings identify an immune braking function and mechanism for murine lncRNAs in inhibiting p65-mediated M1 activation, opening a novel therapeutic avenue of controlling the magnitude of immune responses for HFRS and other inflammatory diseases.
-
Immunology and Microbiology
Stabilization of cytokine mRNAs in iNKT cells requires the serine-threonine kinase IRE1alpha.
In Nature Communications on 17 December 2018 by Govindarajan, S., Gaublomme, D., et al.
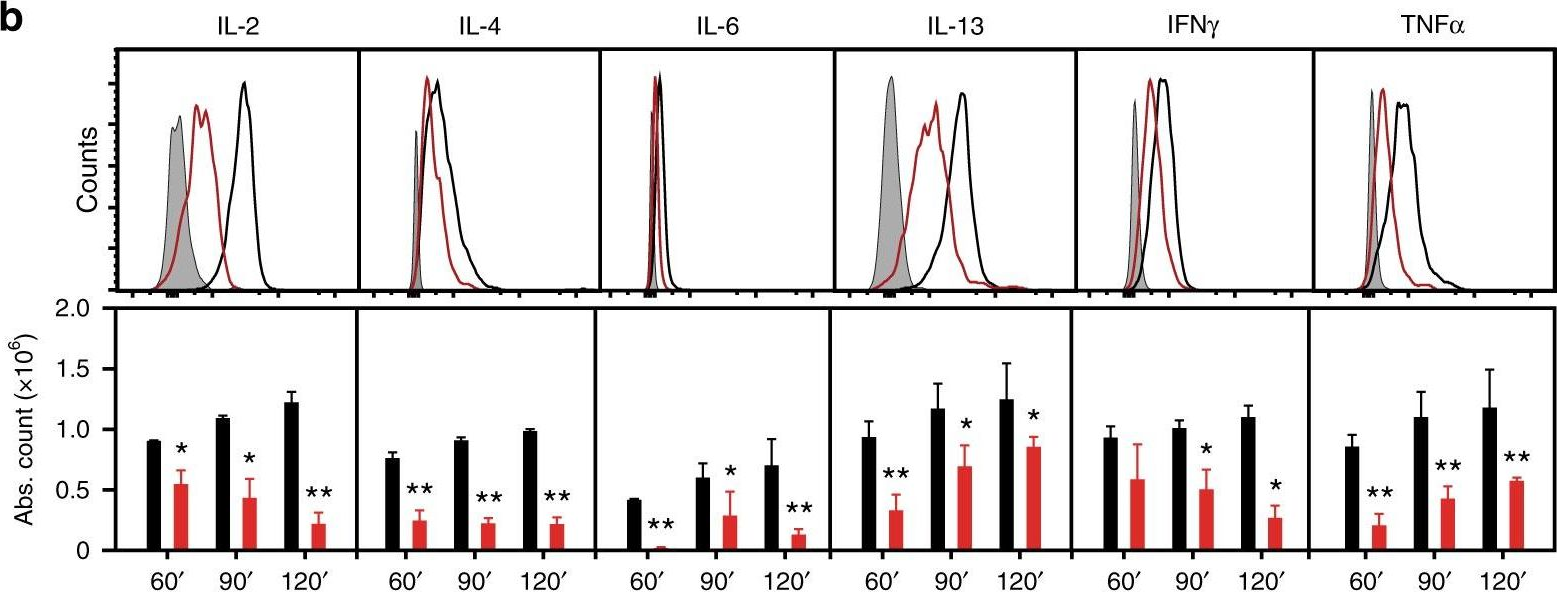
Activated invariant natural killer T (iNKT) cells rapidly produce large amounts of cytokines, but how cytokine mRNAs are induced, stabilized and mobilized following iNKT activation is still unclear. Here we show that an endoplasmic reticulum stress sensor, inositol-requiring enzyme 1α (IRE1α), links key cellular processes required for iNKT cell effector functions in specific iNKT subsets, in which TCR-dependent activation of IRE1α is associated with downstream activation of p38 MAPK and the stabilization of preformed cytokine mRNAs. Importantly, genetic deletion of IRE1α in iNKT cells reduces cytokine production and protects mice from oxazolone colitis. We therefore propose that an IRE1α-dependent signaling cascade couples constitutive cytokine mRNA expression to the rapid induction of cytokine secretion and effector functions in activated iNKT cells.
-
FC/FACS
-
Mus musculus (House mouse)
-
Genetics
In Nat Commun on 17 December 2018 by Govindarajan, S., Gaublomme, D., et al.
Fig.3.B
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 1