Ageing of the immune system is characterized by decreased lymphopoiesis and adaptive immunity, and increased inflammation and myeloid pathologies1,2. Age-related changes in populations of self-renewing haematopoietic stem cells (HSCs) are thought to underlie these phenomena3. During youth, HSCs with balanced output of lymphoid and myeloid cells (bal-HSCs) predominate over HSCs with myeloid-biased output (my-HSCs), thereby promoting the lymphopoiesis required for initiating adaptive immune responses, while limiting the production of myeloid cells, which can be pro-inflammatory4. Ageing is associated with increased proportions of my-HSCs, resulting in decreased lymphopoiesis and increased myelopoiesis3,5,6. Transfer of bal-HSCs results in abundant lymphoid and myeloid cells, a stable phenotype that is retained after secondary transfer; my-HSCs also retain their patterns of production after secondary transfer5. The origin and potential interconversion of these two subsets is still unclear. If they are separate subsets postnatally, it might be possible to reverse the ageing phenotype by eliminating my-HSCs in aged mice. Here we demonstrate that antibody-mediated depletion of my-HSCs in aged mice restores characteristic features of a more youthful immune system, including increasing common lymphocyte progenitors, naive T cells and B cells, while decreasing age-related markers of immune decline. Depletion of my-HSCs in aged mice improves primary and secondary adaptive immune responses to viral infection. These findings may have relevance to the understanding and intervention of diseases exacerbated or caused by dominance of the haematopoietic system by my-HSCs.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Product Citations: 6
Depleting myeloid-biased haematopoietic stem cells rejuvenates aged immunity.
In Nature on 1 April 2024 by Ross, J. B., Myers, L. M., et al.
-
Immunology and Microbiology
-
Stem Cells and Developmental Biology
In Frontiers in Cell and Developmental Biology on 3 August 2021 by Shao, C., Lou, P., et al.
Myoepithelial and luminal cells synergistically expand in the mammary gland during pregnancy, and this process is precisely governed by hormone-related signaling pathways. The bone morphogenetic protein (BMP) signaling pathway is now known to play crucial roles in all organ systems. However, the functions of BMP signaling in the mammary gland remain unclear. Here, we found that BMPR1a is upregulated by hormone-induced Sp1 at pregnancy. Using a doxycycline (Dox)-inducible BMPR1a conditional knockout mouse model, we demonstrated that loss of BMPR1a in myoepithelium results in compromised myoepithelial integrity, reduced mammary stem cells and precocious alveolar differentiation during pregnancy. Mechanistically, BMPR1a regulates the expression of p63 and Slug, two key regulators of myoepithelial maintenance, through pSmad1/5-Smad4 complexes, and consequently activate P-cadherin during pregnancy. Furthermore, we observed that loss of BMPR1a in myoepithelium results in the upregulation of a secreted protein Spp1 that could account for the precocious alveolar differentiation in luminal layer, suggesting a defective basal-to-luminal paracrine signaling mechanism. Collectively, these findings identify a novel role of BMP signaling in maintaining the identity of myoepithelial cells and suppressing precocious alveolar formation.
Copyright © 2021 Shao, Lou, Liu, Bi, Li, Yang, Sheng, Xu, Lv and Yu.
-
FC/FACS
-
Mus musculus (House mouse)
-
Endocrinology and Physiology
TET2 directs mammary luminal cell differentiation and endocrine response.
In Nature Communications on 15 September 2020 by Kim, M. R., Wu, M. J., et al.
Epigenetic regulation plays an important role in governing stem cell fate and tumorigenesis. Lost expression of a key DNA demethylation enzyme TET2 is associated with human cancers and has been linked to stem cell traits in vitro; however, whether and how TET2 regulates mammary stem cell fate and mammary tumorigenesis in vivo remains to be determined. Here, using our recently established mammary specific Tet2 deletion mouse model, the data reveals that TET2 plays a pivotal role in mammary gland development and luminal lineage commitment. We show that TET2 and FOXP1 form a chromatin complex that mediates demethylation of ESR1, GATA3, and FOXA1, three key genes that are known to coordinately orchestrate mammary luminal lineage specification and endocrine response, and also are often silenced by DNA methylation in aggressive breast cancers. Furthermore, Tet2 deletion-PyMT breast cancer mouse model exhibits enhanced mammary tumor development with deficient ERα expression that confers tamoxifen resistance in vivo. As a result, this study elucidates a role for TET2 in governing luminal cell differentiation and endocrine response that underlies breast cancer resistance to anti-estrogen treatments.
-
FC/FACS
-
Mus musculus (House mouse)
-
Endocrinology and Physiology
In Leukemia on 1 August 2020 by Kumar, R., Pereira, R. S., et al.
Therapy resistance in leukemia may be due to cancer cell-intrinsic and/or -extrinsic mechanisms. Mutations within BCR-ABL1, the oncogene giving rise to chronic myeloid leukemia (CML), lead to resistance to tyrosine kinase inhibitors (TKI), and some are associated with clinically more aggressive disease and worse outcome. Using the retroviral transduction/transplantation model of CML and human cell lines we faithfully recapitulate accelerated disease course in TKI resistance. We show in various models, that murine and human imatinib-resistant leukemia cells positive for the oncogene BCR-ABL1T315I differ from BCR-ABL1 native (BCR-ABL1) cells with regards to niche location and specific niche interactions. We implicate a pathway via integrin β3, integrin-linked kinase (ILK) and its role in deposition of the extracellular matrix (ECM) protein fibronectin as causative of these differences. We demonstrate a trend towards a reduced BCR-ABL1T315I+ tumor burden and significantly prolonged survival of mice with BCR-ABL1T315I+ CML treated with fibronectin or an ILK inhibitor in xenogeneic and syngeneic murine transplantation models, respectively. These data suggest that interactions with ECM proteins via the integrin β3/ILK-mediated signaling pathway in BCR-ABL1T315I+ cells differentially and specifically influence leukemia progression. Niche targeting via modulation of the ECM may be a feasible therapeutic approach to consider in this setting.
-
Cancer Research
In Nature Cell Biology on 1 November 2016 by Ni, T., Li, X. Y., et al.
The zinc-finger transcription factor Snail1 is inappropriately expressed in breast cancer and associated with poor prognosis. While interrogating human databases, we uncovered marked decreases in relapse-free survival of breast cancer patients expressing high Snail1 levels in tandem with wild-type, but not mutant, p53. Using a Snail1 conditional knockout model of mouse breast cancer that maintains wild-type p53, we find that Snail1 plays an essential role in tumour progression by controlling the expansion and activity of tumour-initiating cells in preneoplastic glands and established tumours, whereas it is not required for normal mammary development. Growth and survival of preneoplastic as well as neoplastic mammary epithelial cells is dependent on the formation of a Snail1/HDAC1/p53 tri-molecular complex that deacetylates active p53, thereby promoting its proteasomal degradation. Our findings identify Snail1 as a molecular bypass that suppresses the anti-proliferative and pro-apoptotic effects exerted by wild-type p53 in breast cancer.
-
Cancer Research
-
Cell Biology
In Nat Commun on 15 September 2020 by Kim, M. R., Wu, M. J., et al.
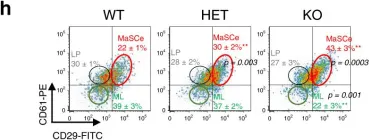
Fig.1.H
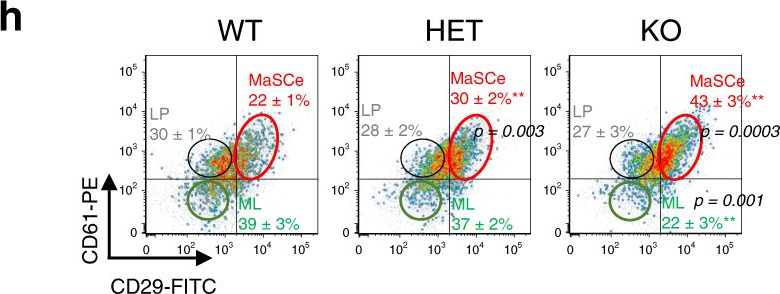
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 1