Platelet integrins, together with other platelet receptors, are known to control hemostasis and thrombosis but also metastatic progression. However, the impact of their exclusive but combined deficiency has never been tested in these processes. In a PF4Cre-β1-/-/β3-/- mouse strain, we found that platelets were exclusively depleted of all integrins. While they displayed impaired binding to fibrinogen and annexin V, P-selectin exposure was normal. Platelet adhesion was abrogated on immobilized fibrinogen and fibrillar fibronectin under shear flow. PF4Cre-β1-/-/β3-/- mice presented an increased bleeding time and a profound defect in experimental models of arterial thrombosis. Platelet adhesion to tumor cells was also reduced, with a pronounced effect on tumor growth and metastatic burden in a model of triple negative breast cancer. Overall, these results confirm the central role of platelet integrins in hemostasis and thrombosis, and highlight their contribution to tumor growth and metastasis formation.
© 2025 The Authors.
Product Citations: 18
Depletion of all platelet integrins impacts hemostasis, thrombosis, and tumor metastasis.
In IScience on 19 September 2025 by Janus-Bell, E., Liboni, C., et al.
-
Cancer Research
In Blood Advances on 12 December 2023 by Ahmad, M. H., Hegde, M., et al.
Germ line mutations in the RUNX1 gene cause familial platelet disorder (FPD), an inherited disease associated with lifetime risk to hematopoietic malignancies (HM). Patients with FPD frequently show clonal expansion of premalignant cells preceding HM onset. Despite the extensive studies on the role of RUNX1 in hematopoiesis, its function in the premalignant bone marrow (BM) is not well-understood. Here, we characterized the hematopoietic progenitor compartments using a mouse strain carrying an FPD-associated mutation, Runx1R188Q. Immunophenotypic analysis showed an increase in the number of hematopoietic stem and progenitor cells (HSPCs) in the Runx1R188Q/+ mice. However, the comparison of Sca-1 and CD86 markers suggested that Sca-1 expression may result from systemic inflammation. Cytokine profiling confirmed the dysregulation of interferon-response cytokines in the BM. Furthermore, the expression of CD48, another inflammation-response protein, was also increased in Runx1R188Q/+ HSPCs. The DNA-damage response activity of Runx1R188Q/+ hematopoietic progenitor cells was defective in vitro, suggesting that Runx1R188Q may promote genomic instability. The differentiation of long-term repopulating HSCs was reduced in Runx1R188Q/+ recipient mice. Furthermore, we found that Runx1R188Q/+ HSPCs outcompete their wild-type counterparts in bidirectional repopulation assays, and that the genetic makeup of recipient mice did not significantly affect the clonal dynamics under this setting. Finally, we demonstrate that Runx1R188Q predisposes to HM in cooperation with somatic mutations found in FPDHM, using 3 mouse models. These studies establish a novel murine FPDHM model and demonstrate that germ line Runx1 mutations induce a premalignant phenotype marked by BM inflammation, selective expansion capacity, defective DNA-damage response, and predisposition to HM.
Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution.
-
Immunology and Microbiology
Serotonin reduction in post-acute sequelae of viral infection.
In Cell on 26 October 2023 by Wong, A. C., Devason, A. S., et al.
Post-acute sequelae of COVID-19 (PASC, "Long COVID") pose a significant global health challenge. The pathophysiology is unknown, and no effective treatments have been found to date. Several hypotheses have been formulated to explain the etiology of PASC, including viral persistence, chronic inflammation, hypercoagulability, and autonomic dysfunction. Here, we propose a mechanism that links all four hypotheses in a single pathway and provides actionable insights for therapeutic interventions. We find that PASC are associated with serotonin reduction. Viral infection and type I interferon-driven inflammation reduce serotonin through three mechanisms: diminished intestinal absorption of the serotonin precursor tryptophan; platelet hyperactivation and thrombocytopenia, which impacts serotonin storage; and enhanced MAO-mediated serotonin turnover. Peripheral serotonin reduction, in turn, impedes the activity of the vagus nerve and thereby impairs hippocampal responses and memory. These findings provide a possible explanation for neurocognitive symptoms associated with viral persistence in Long COVID, which may extend to other post-viral syndromes.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In Nature Cell Biology on 1 January 2023 by Mitchell, C. A., Verovskaya, E. V., et al.
Haematopoietic ageing is marked by a loss of regenerative capacity and skewed differentiation from haematopoietic stem cells (HSCs), leading to impaired blood production. Signals from the bone marrow niche tailor blood production, but the contribution of the old niche to haematopoietic ageing remains unclear. Here we characterize the inflammatory milieu that drives both niche and haematopoietic remodelling. We find decreased numbers and functionality of osteoprogenitors at the endosteum and expansion of central marrow LepR+ mesenchymal stromal cells associated with deterioration of the sinusoidal vasculature. Together, they create a degraded and inflamed old bone marrow niche. Niche inflammation in turn drives the chronic activation of emergency myelopoiesis pathways in old HSCs and multipotent progenitors, which promotes myeloid differentiation and hinders haematopoietic regeneration. Moreover, we show how production of interleukin-1β (IL-1β) by the damaged endosteum acts in trans to drive the proinflammatory nature of the central marrow, with damaging consequences for the old blood system. Notably, niche deterioration, HSC dysfunction and defective regeneration can all be ameliorated by blocking IL-1 signalling. Our results demonstrate that targeting IL-1 as a key mediator of niche inflammation is a tractable strategy to improve blood production during ageing.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
-
FC/FACS
-
Mus musculus (House mouse)
-
Cell Biology
-
Immunology and Microbiology
Stromal inflammation is a targetable driver of hematopoietic aging
Preprint on Research Square on 12 November 2021 by Mitchell, C., Verovskaya, E., et al.
Hematopoietic aging is marked by a loss of regenerative capacity and skewed differentiation from hematopoietic stem cells (HSC) leading to impaired blood production. Signals from the bone marrow (BM) niche tailor blood production, but the contribution of the old niche to hematopoietic aging remains unclear. Here, we characterize the inflammatory milieu that drives both niche and hematopoietic remodeling. We find decreased numbers and functionality of osteoprogenitors (OPr) and expansion of pro-inflammatory perisinusoidal mesenchymal stromal cells (MSC) with deterioration of the sinusoidal vasculature, which together create a degraded and inflamed old BM niche. Niche inflammation, in turn, drives chronic activation of emergency myelopoiesis pathways in old HSCs and multipotent progenitors (MPP), which promotes myeloid differentiation at the expense of lymphoid and erythroid commitment and hinders hematopoietic regeneration. Remarkably, niche deterioration, HSC dysfunction and defective hematopoietic regeneration can all be ameliorated by blocking IL-1 signaling. Our results demonstrate that targeting IL-1 as a key mediator of niche inflammation is a tractable strategy to improve blood production during aging.
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
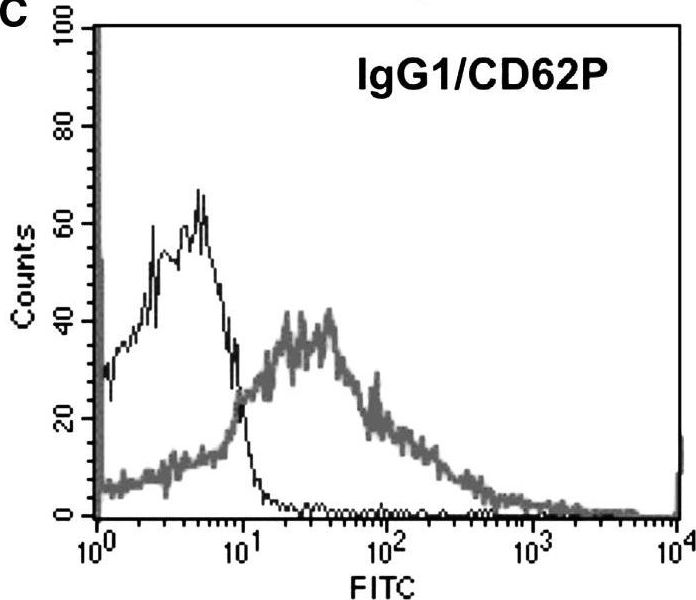
In Biogerontology on 1 August 2015 by Plagg, B., Marksteiner, J., et al.
Fig.1.C
-
FC/FACS
-
Homo sapiens (Human)
Collected and cropped from Biogerontology by CiteAb, provided under a CC-BY license
Image 1 of 1