Current therapeutic options for myelodysplastic neoplasms (MDS)-associated thrombocytopenia are limited. Megakaryocyte maturation might be an innovative therapeutic strategy because its dysregulation profoundly contributes to MDS pathogenesis. Here, we identified crizotinib, a clinically approved anti-cancer drug for anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer, as a potent inducer of megakaryocyte maturation. We demonstrated that crizotinib effectively induced polyploidization to increase the platelet-producing capacity of megakaryocytes derived from an MDS murine model and MDS patients by targeting Aurora kinases rather than its canonical targets, ALK/ROS1/c-MET. Importantly, crizotinib administration substantially ameliorated thrombocytopenia in our preclinical model. Our findings underscore the remarkable potential of crizotinib for drug repurposing and offer a novel therapeutic strategy for MDS patients with thrombocytopenia facing health-related quality of life concerns.
© 2025. The Author(s).
Product Citations: 47
In Leukemia on 1 November 2025 by Kobayashi, H., Komizo, Y., et al.
-
FC/FACS
-
Cancer Research
JMJD3-mediated senescence is required to overcome stress-induced hematopoietic defects.
In EMBO Reports on 1 August 2025 by Nakata, Y., Ueda, T., et al.
Cellular senescence in stem cells compromises regenerative capacity, promotes chronic inflammation, and is implicated in aging. Hematopoietic stem and progenitor cells (HSPCs) are responsible for producing mature blood cells, however, how cellular senescence influences their function is largely unknown. Here, we show that JMJD3, a histone demethylase, activates cellular senescence by upregulating p16Ink4a in competition with Polycomb group proteins, and reprograms HSPC integrity to overcome hematopoietic defects induced by replicative and oncogenic stresses. Jmjd3 deficiency does not alter global H3K27me3 levels, indicating that JMJD3 epigenetically regulates specific and limited JMJD3 targets under stress. JMJD3 deficiency also impairs stem cell potential, proper cell cycle regulation, and WNT pathway activation in HSPCs under stress. These impaired phenotypes are rescued through exogenous and retroviral introduction of p16Ink4a. This JMJD3-p16INK4a axis in hematopoiesis is age-dependent and is distinct from cellular senescence. Treatment with a selective JMJD3 inhibitor attenuates leukemic potential during cellular senescence. Taken together, these results demonstrate that JMJD3-p16INK4a mediates cellular senescence and plays critical roles in the functional integrity of HSPCs under stress.
© 2025. The Author(s).
In eLife on 4 April 2024 by Watanuki, S., Kobayashi, H., et al.
Metabolic pathways are plastic and rapidly change in response to stress or perturbation. Current metabolic profiling techniques require lysis of many cells, complicating the tracking of metabolic changes over time after stress in rare cells such as hematopoietic stem cells (HSCs). Here, we aimed to identify the key metabolic enzymes that define differences in glycolytic metabolism between steady-state and stress conditions in murine HSCs and elucidate their regulatory mechanisms. Through quantitative 13C metabolic flux analysis of glucose metabolism using high-sensitivity glucose tracing and mathematical modeling, we found that HSCs activate the glycolytic rate-limiting enzyme phosphofructokinase (PFK) during proliferation and oxidative phosphorylation (OXPHOS) inhibition. Real-time measurement of ATP levels in single HSCs demonstrated that proliferative stress or OXPHOS inhibition led to accelerated glycolysis via increased activity of PFKFB3, the enzyme regulating an allosteric PFK activator, within seconds to meet ATP requirements. Furthermore, varying stresses differentially activated PFKFB3 via PRMT1-dependent methylation during proliferative stress and via AMPK-dependent phosphorylation during OXPHOS inhibition. Overexpression of Pfkfb3 induced HSC proliferation and promoted differentiated cell production, whereas inhibition or loss of Pfkfb3 suppressed them. This study reveals the flexible and multilayered regulation of HSC glycolytic metabolism to sustain hematopoiesis under stress and provides techniques to better understand the physiological metabolism of rare hematopoietic cells.
© 2023, Watanuki, Kobayashi et al.
-
FC/FACS
-
Mus musculus (House mouse)
-
Stem Cells and Developmental Biology
In IScience on 15 March 2024 by Shi, D., Wang, B., et al.
Pseudouridylation plays a regulatory role in various physiological and pathological processes. A prime example is the mitochondrial myopathy, lactic acidosis, and sideroblastic anemia syndrome (MLASA), characterized by defective pseudouridylation resulting from genetic mutations in pseudouridine synthase 1 (PUS1). However, the roles and mechanisms of pseudouridylation in normal erythropoiesis and MLASA-related anemia remain elusive. We established a mouse model carrying a point mutation (R110W) in the enzymatic domain of PUS1, mimicking the common mutation in human MLASA. Pus1-mutant mice exhibited anemia at 4 weeks old. Impaired mitochondrial oxidative phosphorylation was also observed in mutant erythroblasts. Mechanistically, mutant erythroblasts showed defective pseudouridylation of targeted tRNAs, altered tRNA profiles, decreased translation efficiency of ribosomal protein genes, and reduced globin synthesis, culminating in ineffective erythropoiesis. Our study thus provided direct evidence that pseudouridylation participates in erythropoiesis in vivo. We demonstrated the critical role of pseudouridylation in regulating tRNA homeostasis, cytoplasmic translation, and erythropoiesis.
© 2024 The Authors.
-
Biochemistry and Molecular biology
-
Cell Biology
-
Genetics
In eLife on 11 December 2023 by Yao, L., Lu, J., et al.
Insufficient bone fracture repair represents a major clinical and societal burden and novel strategies are needed to address it. Our data reveal that the transforming growth factor-β superfamily member Activin A became very abundant during mouse and human bone fracture healing but was minimally detectable in intact bones. Single-cell RNA-sequencing revealed that the Activin A-encoding gene Inhba was highly expressed in a unique, highly proliferative progenitor cell (PPC) population with a myofibroblast character that quickly emerged after fracture and represented the center of a developmental trajectory bifurcation producing cartilage and bone cells within callus. Systemic administration of neutralizing Activin A antibody inhibited bone healing. In contrast, a single recombinant Activin A implantation at fracture site in young and aged mice boosted: PPC numbers; phosphorylated SMAD2 signaling levels; and bone repair and mechanical properties in endochondral and intramembranous healing models. Activin A directly stimulated myofibroblastic differentiation, chondrogenesis and osteogenesis in periosteal mesenchymal progenitor culture. Our data identify a distinct population of Activin A-expressing PPCs central to fracture healing and establish Activin A as a potential new therapeutic tool.
© 2023, Yao et al.
-
FC/FACS
-
Mus musculus (House mouse)
In Cancers (Basel) on 28 July 2020 by Kuziel, G., Thompson, V., et al.
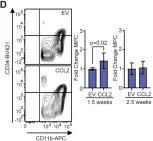
Fig.4.D
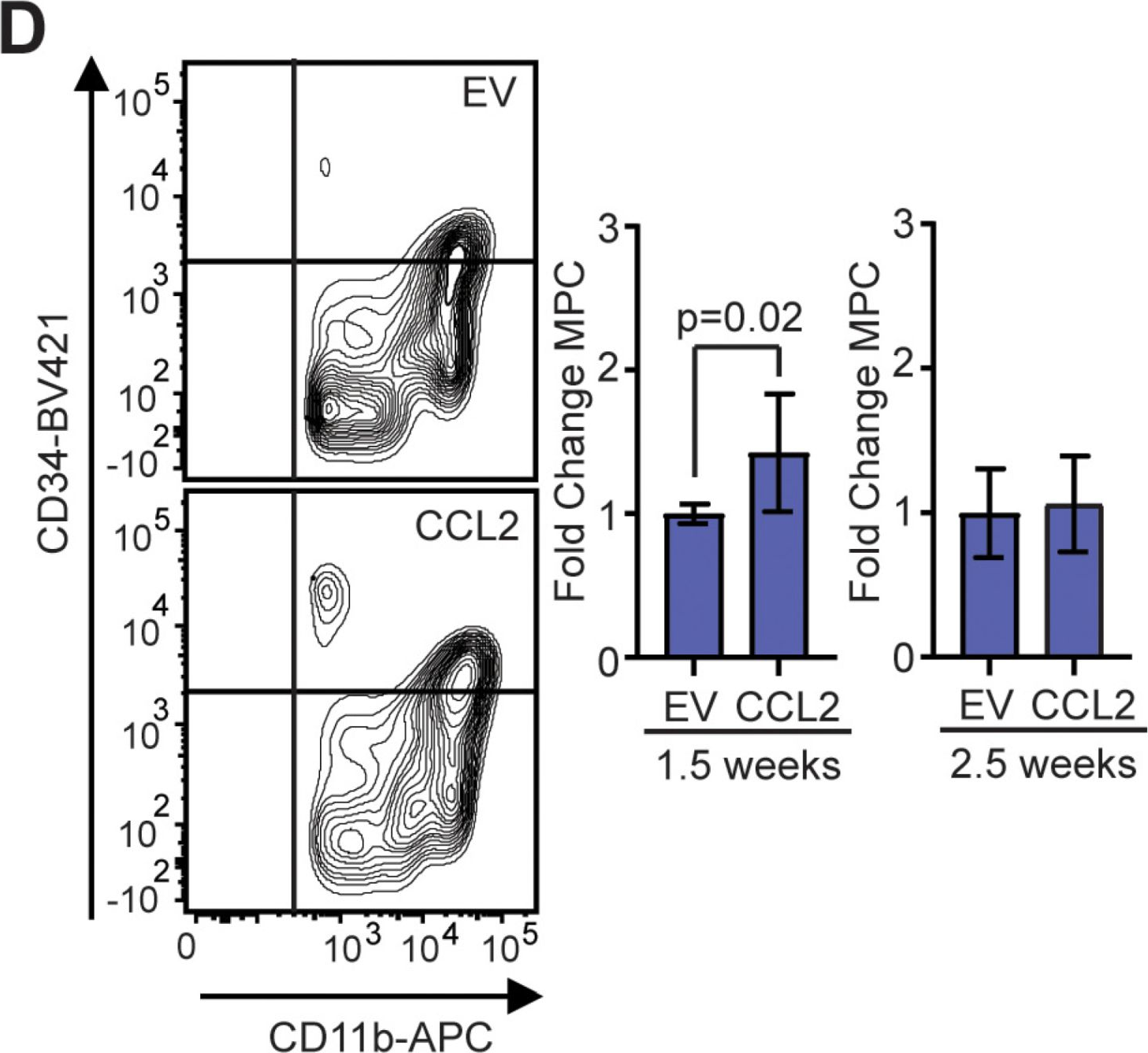
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Cancers by CiteAb, provided under a CC-BY license
Image 1 of 3
In Cancers (Basel) on 28 July 2020 by Kuziel, G., Thompson, V., et al.
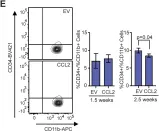
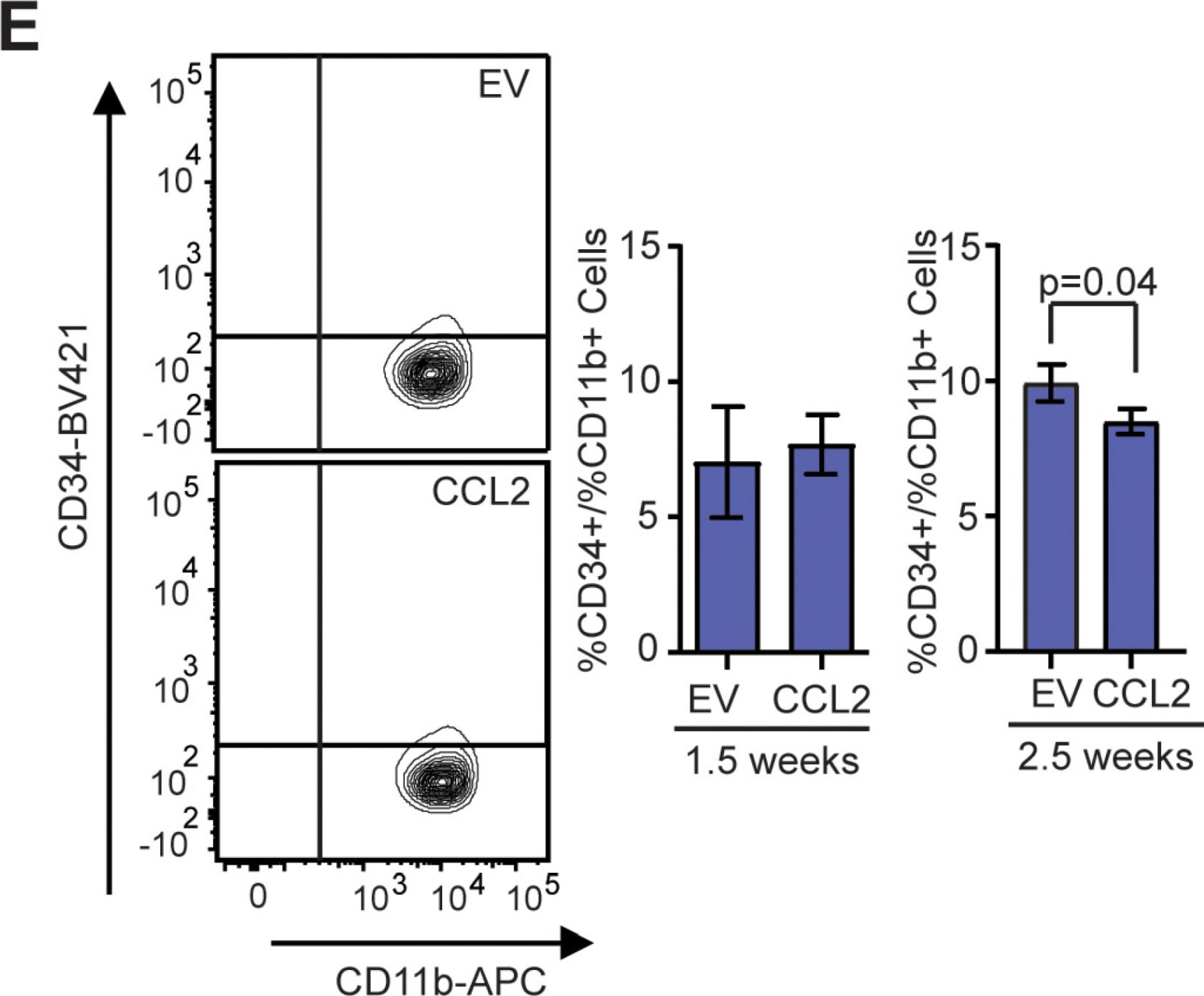
Fig.4.E
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Cancers by CiteAb, provided under a CC-BY license
Image 1 of 3
In Nat Commun on 6 April 2016 by Yosef, R., Pilpel, N., et al.
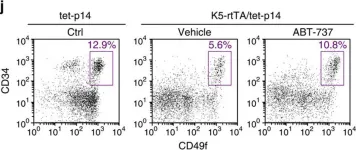
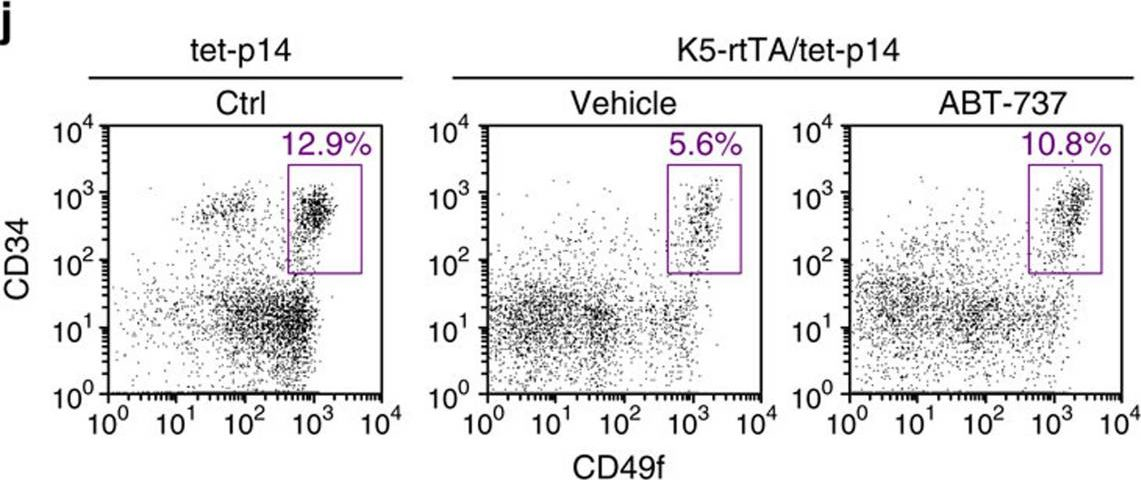
Fig.5.J
-
FC/FACS
-
Mus musculus (House mouse)
Collected and cropped from Nature Communications by CiteAb, provided under a CC-BY license
Image 1 of 3