Itaconate, a macrophage-specific anti-inflammatory metabolite, has recently emerged as a critical regulator in rheumatoid arthritis pathogenesis. We found that itaconate is a TNF-α responsive metabolite significantly elevated in the serum and synovial fluid of rheumatoid arthritis patients and we demonstrated that itaconate is primarily produced by inflammatory macrophages rather than osteoclasts or osteoblasts. In TNF-transgenic and Irg1-/- hybrid mice, a more severe bone destruction phenotype was observed. Administration of itaconate prevents excessive activation of osteoclasts by inhibiting Tet2 enzyme activity. Furthermore, exogenous administration of itaconate or its derivative, 4-octyl-itaconate, inhibits arthritis progression and mitigates bone destruction, offering a potential therapeutic strategy for rheumatoid arthritis. This study elucidates that TNF-α drives macrophage-derived itaconate production to epigenetically suppress osteoclast hyperactivation through Tet2 inhibition, establishing itaconate and its derivative OI as novel therapeutic agents against rheumatoid arthritis -associated bone destruction.
© 2025. The Author(s).
Product Citations: 97
In Bone Research on 11 June 2025 by Rong, K., Wang, D., et al.
-
Genetics
-
Immunology and Microbiology
In Diabetes Metabolism Journal on 22 May 2025 by Song, D. G., Pak, S., et al.
Obesity is a rapidly increasing global health issue, which is associated with glucose and insulin resistance. Phosphodiesterase type 5 (PDE5) inhibitors (PDE5i) are known for their ability to enhance blood flow and vascular stability and are widely used to treat conditions such as erectile dysfunction, pulmonary hypertension, heart failure, and cancer. However, studies investigating the role of PDE5i in alleviating obesity and metabolic diseases remains unclear. Therefore, we investigated the effects of PDE5i on obesity and metabolic disorders in diet-induced obese mice and its underlying mechanisms.
PDE5i was administered to high-fat diet (HFD)-fed C57BL/6J mice for 6 to 7 weeks. Body weight and food intake were measured weekly, and baseline metabolic rates, physical activity, and glucose and insulin tolerance tests were assessed during PDE5i administration. Macrophages and T-cells in the gonadal white adipose tissue (gWAT) were analyzed by flow cytometry. Vascular stability and blood flow in gWAT were analyzed via immunostaining and in vivo live imaging. RAW264.7 cells and bone marrow-derived macrophages were used to determine immunoregulatory effects of PDE5i.
In HFD-fed mice, PDE5i administration significantly enhanced systemic insulin sensitivity and AKT phosphorylation in gWAT. PDE5i reduced the M1/M2 ratio of gWAT macrophages of obese mice. These phenomena were associated with enhanced blood flow to the gWAT. In vitro experiments revealed that PDE5i suppressed lipopolysaccharide-induced proinflammatory cytokine production and increased the mRNA expression of genes associated with M2 polarization.
PDE5i plays a role in regulating adipose tissue inflammation and thus holds promise as a therapeutic agent for metabolic enhancement.
-
Endocrinology and Physiology
-
Immunology and Microbiology
In International Journal of Biological Sciences on 19 May 2025 by Bian, Z., Chen, J., et al.
Mutations in TP53, particularly the p.R248Q variant, contribute to the progression of castration-resistant prostate cancer (CRPC) by reshaping the tumor microenvironment (TME). This study examined the impact of p.R248Q (mutp53) on immune suppression and CRPC progression. We introduced the Trp53 p.R245Q mutation into RM-1 mouse prostate cancer (PCa) cells via CRISPR/Cas9, which mimics human TP53 p.R248Q. These cells were implanted into C57BL/6 mice to model tumor progression and immune interactions. Mice were treated with JAK2 and STAT3 inhibitors to assess immune and tumor responses. Tumor behavior and immune responses were analyzed via histology, immunofluorescence, flow cytometry, Enzyme-linked immunosorbent assay (ELISA), and bioinformatics. Findings were validated in the C4-2 human PCa cell line. Compared with wild-type p53, TP53 mutations were present in 27% of PCa patients and were significantly correlated with reduced overall survival (p < 0.001, HR = 1.97) and recurrence-free survival (p = 0.02, HR = 1.62). The p.R248Q mutation was most prevalent. Gene-edited mutp53 cells exhibited increased proliferation and tumorigenicity. Screening and validation confirmed that IL6/JAK2/STAT3 pathway activation in mutp53 tumors led to immune microenvironment alterations. Flow cytometry and immunofluorescence revealed an immunosuppressive profile, with decreased proinflammatory cytokines and elevated anti-inflammatory factors. Coimmunoprecipitation revealed that mutp53 competes with SHP1 for STAT3 binding, sustaining its activation. Inhibition of STAT3 reduced mutp53-driven immune suppression and tumor progression. Mutp53 promotes an immunosuppressive TME and facilitates CRPC progression through the STAT3 pathway, underscoring its potential as a therapeutic target.
© The author(s).
-
Cancer Research
Macrophage PKM2 depletion ameliorates hepatic inflammation and acute liver injury in mice.
In Frontiers in Pharmacology on 12 May 2025 by Kang, Z., Xie, R., et al.
Pyruvate kinase M2 (PKM2), the rate-limiting enzyme of glycolysis, plays a critical role in macrophage activation and a broad spectrum of chronic liver diseases. However, whether PKM2 contributes to the pathogenesis of acute liver injury (ALI) remains largely unexplored.
PKM2 expression was assessed in human and mouse ALI livers. Macrophage-specific PKM2 knockout mice were challenged by two independent ALI models, induced by acetaminophen (APAP) and lipopolysaccharide/D-galactosamine (LPS/D-GalN), to explore the role and regulatory mechanism of macrophage PKM2 in ALI progression.
By bioinformatic screening and analysis of ALI liver, we found that PKM2 was significantly upregulated in the liver tissues of ALI patients and mice. Immunofluorescence staining further demonstrated that PKM2 was markedly upregulated in macrophages during ALI progression. Notably, macrophage PKM2 depletion effectively alleviated APAP- and LPS/D-GalN-induced ALI, as demonstrated by ameliorated immune cells infiltration, pro-inflammatory mediators, and hepatocellular cell death. PKM2-deficient macrophages showed M2 anti-inflammatory polarization in vivo and in vitro. Furthermore, PKM2 deletion limited HIF-1α signaling and aerobic glycolysis of macrophages, which thereby attenuated macrophage pro-inflammatory activation and hepatocyte injury. Pharmacological PKM2 antagonist efficiently ameliorated liver injury and prolonged the survival of mice in APAP-induced ALI model.
Our study highlights the pivotal role of macrophage PKM2 in advancing ALI, and therapeutic targeting of PKM2 may serve as a novel strategy to combat ALI.
Copyright © 2025 Kang, Xie, Cui, Chen, Xie, Li, Lv, Ye, Zhao, Zhang, Hong and Qu.
-
FC/FACS
-
Mus musculus (House mouse)
-
Immunology and Microbiology
-
Pharmacology
In JCI Insight on 8 May 2025 by Xu, S., Li, H., et al.
The inflammatory response after myocardial infarction (MI) is a precisely regulated process that greatly affects subsequent wound healing and remodeling. However, understanding about the process is still limited. Macrophages are critically involved in inflammation resolution after MI. Krüppel-like factor 9 (Klf9) is a C2H2 zinc finger-containing transcription factor that has been implicated in glucocorticoid regulation of macrophages. However, the contribution of Klf9 to macrophage phenotype and function in the context of MI remains unclear. Our study revealed that KLF9 deficiency resulted in higher mortality and cardiac rupture rate, as well as a considerable exacerbation in cardiac function. Single-cell RNA sequencing and flow cytometry analyses revealed that, compared with WT mice, Klf9-/- mice displayed excessive neutrophil infiltration, insufficient macrophage infiltration, and a reduced proportion of monocyte-derived CD206+ macrophages after MI. Moreover, the expression of IFN-γ/STAT1 pathway genes in Klf9-/- cardiac macrophages was dysregulated, characterized by insufficient expression at 1 day post-MI and excessive expression at day 3 post-MI. Mechanistically, Klf9 directly binds to the promoters of Stat1 gene, regulating its transcription. Overall, these findings indicate that Klf9 beneficially influences wound healing after MI by modulating macrophage recruitment and differentiation by regulating the IFN-γ/STAT1 signaling pathway.
-
FC/FACS
-
Mus musculus (House mouse)
-
Cardiovascular biology
-
Immunology and Microbiology
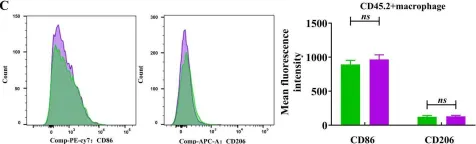
In PLoS Negl Trop Dis on 1 May 2025 by Zhang, B., Li, X., et al.
Fig.5.C

-
FC/FACS
-
Collected and cropped from PLoS Negl Trop Dis by CiteAb, provided under a CC-BY license
Image 1 of 2
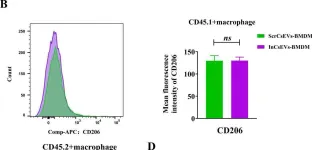
In PLoS Negl Trop Dis on 1 May 2025 by Zhang, B., Li, X., et al.
Fig.5.B

-
FC/FACS
-
Collected and cropped from PLoS Negl Trop Dis by CiteAb, provided under a CC-BY license
Image 1 of 2