Integrins provide an essential bridge between cancer cells and the extracellular matrix, playing a central role in every stage of disease progression. Despite the recognized importance of integrin phosphorylation in several biological processes, the regulatory mechanisms and their relevance remained elusive. Here we engineer a fluorescence resonance energy transfer biosensor for integrin β1 phosphorylation, screening 96 protein tyrosine phosphatases and identifying Shp2 and PTP-PEST as negative regulators to address this gap. Mutation of the integrin NPxY(783/795) sites revealed the importance of integrin phosphorylation for efficient cancer cell invasion, further supported by inhibition of the identified integrin phosphorylation regulators Shp2 and Src kinase. Using proteomics approaches, we uncovered Cofilin as a component of the phosphorylated integrin-Dok1 complex and linked this axis to effective invadopodia formation, a process supporting breast cancer invasion. These data further implicate dynamic modulation of integrin β1 phosphorylation at NPxY sites at different stages of metastatic dissemination.
© 2025. The Author(s).
Product Citations: 567
Dynamic regulation of integrin β1 phosphorylation supports invasion of breast cancer cells.
In Nature Cell Biology on 26 May 2025 by Conway, J. R., Joshi, O., et al.
-
WB
-
Cancer Research
-
Cell Biology
MuSK is a substrate for CaMK2β but this interaction is dispensable for MuSK activation in vivo.
In Scientific Reports on 28 April 2025 by Prömer, J. J., Wolske, S., et al.
The neuromuscular junction (NMJ) is the unique interface between lower motor neurons and skeletal muscle fibers and is indispensable for muscle function. Tight control of its localized formation at the center of every muscle fiber, and maintenance throughout lifetime are sustained by muscle-specific kinase (MuSK). MuSK acts as central regulator of acetylcholine receptor clustering at the postsynapse. Localized and temporally controlled signaling of MuSK is primarily achieved by tyrosine autophosphorylation and inhibition thereof. Previous investigations suggested serine phosphorylation of the activation domain as an additional modulator of MuSK activation. Here we identified calcium/calmodulin dependent protein kinase II (CaMK2) and in particular CaMK2β as novel catalyst of MuSK activation and confirmed its capability to phosphorylate MuSK in heterologous cells. However, whereas CaMK2β absence in muscle cells reduced AChR clustering, MuSK phosphorylation was unchanged. Accordingly, we ruled out MuSK phosphorylation as the cause of synapse fragmentation in a mouse model for myotonic dystrophy type 1, in which the muscle-specific splice-variant of CaMK2β is missing, or as the cause of ataxia or delayed muscle development in CaMK2β knockout animals. Histological characterization of muscles of CaMK2β knockout mice indicated specific roles of CaMK2β in fast glycolytic versus slow oxidative muscle. Taken together, our data shows that MuSK can be phosphorylated by CaMK2β, but loss of CaMK2β is likely compensated by other CaMK2 paralogs at the NMJ.
© 2025. The Author(s).
In Cell Communication and Signaling : CCS on 7 January 2025 by Wang, K., Qu, H., et al.
Polymerase delta-interacting protein 2 (Poldip2) is a novel regulator of vascular permeability that has been shown to be involved in aggravating blood-brain barrier (BBB) disruption following stroke; however, the underlying mechanisms are unknown. While endothelial tight junctions (TJ) are critical mediators of BBB permeability, the effect of Poldip2 on TJ function has not been elucidated yet. Here, we aim to define the mechanism by which Poldip2 mediates BBB disruption, specifically focusing on phosphorylation and stabilization of the TJ integral protein ZO-1.
Cerebral ischemia was induced in endothelial-specific Poldip2 knockout mice and controls. Cerebral vascular permeability was assessed by Evans blue dye extravasation. Endothelial-specific Poldip2 deletion abolished Evans blue dye extravasation after ischemia induction. In vitro permeability assays demonstrated that Poldip2 knockdown suppressed TNF-α-induced endothelial cell (EC) permeability. Immunofluorescence staining showed that Poldip2 depletion prevented TNF-α-induced ZO-1 disruption at interendothelial junctions. Conversely, Poldip2 overexpression increased endothelial permeability, loss of ZO-1 localization at cell-cell junctions and enhanced reactive oxygen species (ROS) production. Treatment with the antioxidant N-acetyl cysteine (NAC) reduced Poldip2-induced ZO-1 disruption at inter interendothelial junctions. Immunoprecipitation studies demonstrated Poldip2 overexpression induced tyrosine phosphorylation of ZO-1, which was prevented by treatment with NAC or MitoTEMPO, a mitochondrial ROS scavenger.
These data reveal a novel mitochondrial ROS-driven mechanism by which Poldip2 induces ZO-1 tyrosine phosphorylation and promotes EC permeability following cerebral ischemia.
© 2025. The Author(s).
-
Endocrinology and Physiology
Neoplastic ICAM-1 protects lung carcinoma from apoptosis through ligation of fibrinogen.
In Cell Death & Disease on 21 August 2024 by Wang, S., Wang, J., et al.
Intercellular cell adhesion molecule-1 (ICAM-1) is frequently overexpressed in non-small cell lung cancer (NSCLC) and associated with poor prognosis. However, the mechanism underlying the negative effects of neoplastic ICAM-1 remains obscure. Herein, we demonstrate that the survival of NSCLC cells but not normal human bronchial epithelial cells requires an anti-apoptosis signal triggered by fibrinogen γ chain (FGG)-ICAM-1 interaction. ICAM-1-FGG ligation preserves the tyrosine phosphorylation of ICAM-1 cytoplasmic domain and its association with SHP-2, and subsequently promotes Akt and ERK1/2 activation but suppresses JNK and p38 activation. Abolishing ICAM-1-FGG interaction induces NSCLC cell death by activating caspase-9/3 and significantly inhibits tumor development in a mouse xenograft model. Finally, we developed a monoclonal antibody against ICAM-1-FGG binding motif, which blocks ICAM-1‒FGG interaction and effectively suppresses NSCLC cell survival in vitro and tumor growth in vivo. Thus, suppressing ICAM-1-FGG axis provides a potential strategy for NSCLC targeted therapy.
© 2024. The Author(s).
-
Cancer Research
-
Cell Biology
In Biomolecules on 2 December 2023 by Kioumourtzoglou, D., Black, H. L., et al.
A major consequence of insulin binding its receptor on fat and muscle cells is the stimulation of glucose transport into these tissues. This is achieved through an increase in the exocytic trafficking rate of the facilitative glucose transporter GLUT4 from intracellular stores to the cell surface. Delivery of GLUT4 to the cell surface requires the formation of functional SNARE complexes containing Syntaxin 4, SNAP23, and VAMP2. Insulin stimulates the formation of these complexes and concomitantly causes phosphorylation of Syntaxin 4. Here, we use a combination of biochemistry and cell biological approaches to provide a mechanistic link between these observations. We present data to support the hypothesis that Tyr-115 and Tyr-251 of Syntaxin 4 are direct substrates of activated insulin receptors, and that these residues modulate the protein's conformation and thus regulate the rate at which Syntaxin 4 forms SNARE complexes that deliver GLUT4 to the cell surface. This report provides molecular details on how the cell regulates SNARE-mediated membrane traffic in response to an external stimulus.
-
Biochemistry and Molecular biology
-
Endocrinology and Physiology
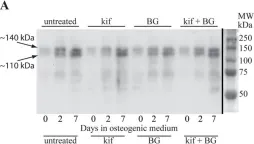
In J Cell Sci on 14 February 2018 by Wilson, K. M., Jagger, A. M., et al.
Fig.5.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Cell Sci by CiteAb, provided under a CC-BY license
Image 1 of 4
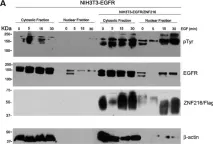
In Oncotarget on 15 November 2016 by Mincione, G., Di Marcantonio, M. C., et al.
Fig.6.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 4
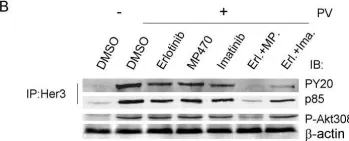
In BMC Cancer on 11 May 2009 by Qi, W., Cooke, L. S., et al.
Fig.6.B

-
WB
-
Collected and cropped from BMC Cancer by CiteAb, provided under a CC-BY license
Image 1 of 4
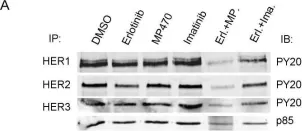
In BMC Cancer on 11 May 2009 by Qi, W., Cooke, L. S., et al.
Fig.6.A

-
WB
-
Collected and cropped from BMC Cancer by CiteAb, provided under a CC-BY license
Image 1 of 4