A bstract Brain-derived neurotrophic factor (BDNF) is essential for neuronal survival, differentiation, and plasticity. In epilepsy, BDNF exhibits a dual role, exerting both antiepileptic and pro-epileptic effects. The cleavage of its main receptor, full-length tropomyosin-related kinase B (TrkB-FL), was suggested to occur in status epilepticus (SE) in vitro. Moreover, under excitotoxic conditions, TrkB-FL was found to be cleaved, resulting in the formation of a new intracellular fragment, TrkB-ICD. Thus, we hypothesized that TrkB-FL cleavage and TrkB-ICD formation could represent an uncovered mechanism in epilepsy. We used a rat model of mesial temporal lobe epilepsy (mTLE) induced by kainic acid (KA) to investigate TrkB-FL cleavage and TrkB-ICD formation during SE and established epilepsy (EE). Animals treated with 10 mg/kg of KA exhibited TrkB-FL cleavage during SE, with hippocampal levels of TrkB-FL and TrkB-ICD correlating with seizure severity. Notably, TrkB-FL cleavage and TrkB-ICD formation were also detected in animals with EE, which exhibited spontaneous recurrent convulsive seizures, neuronal death, mossy fiber sprouting, and long-term memory impairment. Importantly, hippocampal samples from patients with refractory epilepsy also showed TrkB-FL cleavage with increased TrkB-ICD levels. Additionally, overexpression of TrkB-ICD in the hippocampus of healthy rodents resulted in long-term memory impairment. Our findings suggest that TrkB-FL cleavage and the subsequent TrkB-ICD production occur throughout epileptogenesis, with the extent of cleavage correlating positively with seizure occurrence. Moreover, we found that TrkB-ICD impairs memory. This work uncovers a novel mechanism in epileptogenesis that could serve as a potential therapeutic target in mTLE, with implications for preserving cognitive function.
Product Citations: 62
Preprint on BioRxiv : the Preprint Server for Biology on 20 December 2024 by Ribeiro-Rodrigues, L., Fonseca-Gomes, J., et al.
In British Journal of Pharmacology on 1 September 2024 by Wang, D., Li, X., et al.
Traumatic brain injury (TBI) causes lifelong physical and psychological dysfunction in affected individuals. The current study investigated the effects of chronic nicotine exposure via E-cigarettes (E-cig) (vaping) on TBI-associated behavioural and biochemical changes.
Adult C57/BL6J male mice were subjected to controlled cortical impact (CCI) followed by daily exposure to E-cig vapour for 6 weeks. Sensorimotor functions, locomotion, and sociability were subsequently evaluated by nesting, open field, and social approach tests, respectively. Immunoblots were conducted to examine the expression of mature brain-derived neurotrophic factor (mBDNF) and associated downstream proteins (p-Erk, p-Akt). Histological analyses were performed to evaluate neuronal survival and neuroinflammation.
Post-injury chronic nicotine exposure significantly improved nesting performance in CCI mice. Histological analysis revealed increased survival of cortical neurons in the perilesion cortex with chronic nicotine exposure. Immunoblots revealed that chronic nicotine exposure significantly up-regulated mBDNF, p-Erk and p-Akt expression in the perilesion cortex of CCI mice. Immunofluorescence microscopy indicated that elevated mBDNF and p-Akt expression were mainly localized within cortical neurons. Immunolabelling of Iba1 demonstrated that chronic nicotine exposure attenuated microglia-mediated neuroinflammation.
Post-injury chronic nicotine exposure via vaping facilitates recovery of sensorimotor function by upregulating neuroprotective mBDNF/TrkB/Akt/Erk signalling. These findings suggest potential neuroprotective properties of nicotine despite its highly addictive nature. Thus, understanding the multifaceted effects of chronic nicotine exposure on TBI-associated symptoms is crucial for paving the way for informed and properly managed therapeutic interventions.
© 2024 The Authors. British Journal of Pharmacology published by John Wiley & Sons Ltd on behalf of British Pharmacological Society.
-
Pharmacology
In Frontiers in Neuroscience on 23 August 2024 by Peterson, L., Nguyen, J., et al.
Environmental enrichment combined with the glycine transporter-1 inhibitor Org24598 (EE+ORG) during cocaine-cue extinction (EXT) inhibited reacquisition of 1.0 mg/kg cocaine self-administration in male but not female rats in a previous investigation. In this investigation, we determined if this treatment benefit in males required EXT training and ascertained the molecular basis for the observed sex difference in treatment efficacy. Nine groups of male rats trained to self-administer 1.0 mg/kg cocaine or receiving yoked-saline underwent EXT or NoEXT with or without EE and/or ORG. Next, they underwent reacquisition of cocaine self-administration or were sacrificed for molecular analysis of 9 protein targets indicative of neuroplasticity in four brain regions. Two groups of female rats trained to self-administer 1.0 mg/kg cocaine also underwent EXT with or without EE + ORG and were sacrificed for molecular analysis, as above. EE + ORG facilitated the rate of EXT learning in both sexes, and importantly, the therapeutic benefit of EE + ORG for inhibiting cocaine relapse required EXT training. Males were more sensitive than females to neuroplasticity-inducing effects of EE + ORG, which prevented reductions in total GluA1 and PSD95 proteins selectively in basolateral amygdala of male rats trained to self-administer cocaine and receiving EXT. Females were deficient in expression of multiple protein targets, especially after EE + ORG. These included total GluA1 and PSD95 proteins in basolateral amygdala, and total TrkB protein in basolateral amygdala, dorsal hippocampus, and ventromedial prefrontal cortex. Together, these results support the clinical view that sex-specific pharmacological and behavioral treatment approaches may be needed during cue exposure therapy to inhibit cocaine relapse.
Copyright © 2024 Peterson, Nguyen, Ghani, Rodriguez-Echemendia, Qiao, Guwn, Man and Kantak.
-
Rattus norvegicus (Rat)
-
Neuroscience
In The Journal of Biological Chemistry on 1 August 2024 by Ma, L., Kasula, R. K., et al.
Mutations in the endosomal Na+/H+ exchanger 6 (NHE6) cause Christianson syndrome, an X-linked neurological disorder. NHE6 functions in regulation of endosome acidification and maturation in neurons. Using yeast two-hybrid screening with the NHE6 carboxyl terminus as bait, we identify Golgi-associated, gamma adaptin ear-containing, ADP-ribosylation factor (ARF) binding protein 1 (GGA1) as an interacting partner for NHE6. We corroborated the NHE6-GGA1 interaction using: coimmunoprecipitation; overexpressed constructs in mammalian cells; and coimmunoprecipitation of endogenously expressed GGA1 and NHE6 from neuroblastoma cells, as well as from the mouse brain. We demonstrate that GGA1 interacts with organellar NHEs (NHE6, NHE7, and NHE9) and that there is significantly less interaction with cell-surface localized NHEs (NHE1 and NHE5). By constructing hybrid NHE1/NHE6 exchangers, we demonstrate the cytoplasmic tail of NHE6 interacts most strongly with GGA1. We demonstrate the colocalization of NHE6 and GGA1 in cultured, primary hippocampal neurons, using super-resolution microscopy. We test the hypothesis that the interaction of NHE6 and GGA1 functions in the localization of NHE6 to the endosome compartment. Using subcellular fractionation experiments, we show that NHE6 is mislocalized in GGA1 KO cells, wherein we find less NHE6 in endosomes, but more NHE6 transport to lysosomes, and more Golgi retention of NHE6, with increased exocytosis to the surface plasma membrane. Consistent with NHE6 mislocalization, and Golgi retention, we find the intraluminal pH in Golgi to be alkalinized in GGA1-null cells. Our study demonstrates a new interaction between NHE6 and GGA1 which functions in the localization of this intracellular NHE to the endosome compartment.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
-
Biochemistry and Molecular biology
-
Cell Biology
In Proceedings of the National Academy of Sciences of the United States of America on 23 April 2024 by Wang, C. S., McCarthy, C. I., et al.
Brain-derived neurotrophic factor (BDNF) plays a critical role in synaptic physiology, as well as mechanisms underlying various neuropsychiatric diseases and their treatment. Despite its clear physiological role and disease relevance, BDNF's function at the presynaptic terminal, a fundamental unit of neurotransmission, remains poorly understood. In this study, we evaluated single synapse dynamics using optical imaging techniques in hippocampal cell cultures. We find that exogenous BDNF selectively increases evoked excitatory neurotransmission without affecting spontaneous neurotransmission. However, acutely blocking endogenous BDNF has no effect on evoked or spontaneous release, demonstrating that different approaches to studying BDNF may yield different results. When we suppressed BDNF-Tropomyosin receptor kinase B (TrkB) activity chronically over a period of days to weeks using a mouse line enabling conditional knockout of TrkB, we found that evoked glutamate release was significantly decreased while spontaneous release remained unchanged. Moreover, chronic blockade of BDNF-TrkB activity selectively downscales evoked calcium transients without affecting spontaneous calcium events. Via pharmacological blockade by voltage-gated calcium channel (VGCC) selective blockers, we found that the changes in evoked calcium transients are mediated by the P/Q subtype of VGCCs. These results suggest that BDNF-TrkB activity increases presynaptic VGCC activity to selectively increase evoked glutamate release.
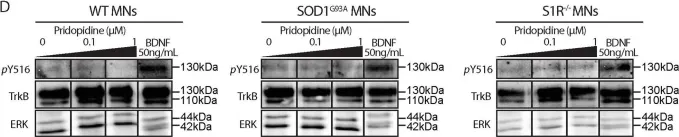
In Cell Death Dis on 1 March 2019 by Ionescu, A., Gradus, T., et al.
Fig.5.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 3
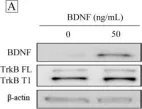
In PLoS One on 13 June 2017 by Tsai, Y. F., Tseng, L. M., et al.
Fig.3.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 3
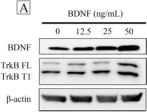
In PLoS One on 13 June 2017 by Tsai, Y. F., Tseng, L. M., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 3