
Type I interferon (IFN) signalling induces the expression of several hundred IFN-stimulated genes (ISGs) that provide an unfavourable environment for viral replication. To prevent an overexuberant response and autoinflammatory disease, IFN signalling requires tight control. One critical regulator is the ubiquitin-like protein IFN-stimulated gene 15 (ISG15), evidenced by autoinflammatory disease in patients with inherited ISG15 deficiencies. Current models suggest that ISG15 stabilises ubiquitin-specific peptidase 18 (USP18), a well-established negative regulator of IFN signalling. USP18 also functions as an ISG15-specific peptidase that cleaves ISG15 from ISGylated proteins; however, USP18's catalytic activity is dispensable for controlling IFN signalling. Here, we show that the ISG15-dependent stabilisation of USP18 involves hydrophobic interactions reliant on tryptophan 123 (W123) in ISG15. Nonetheless, while USP18 stabilisation is necessary, it is not sufficient for the regulation of IFN signalling; ISG15 C-terminal mutants with significantly reduced affinity still stabilised USP18, yet the magnitude of signalling resembled ISG15-deficient cells. Hence, USP18 requires non-covalent interactions with the ISG15 C-terminal diGlycine motif to promote its regulatory function. It shows ISG15 is a repressor of type I IFN signalling beyond its role as a USP18 stabiliser.
© 2025 The Author(s). European Journal of Immunology published by Wiley‐VCH GmbH.
Product Citations: 31
In European Journal of Immunology on 1 February 2025 by Vasou, A., Nightingale, K., et al.
-
Immunology and Microbiology
Preprint on BioRxiv : the Preprint Server for Biology on 13 September 2024 by Prus, W., Grabowski, F., et al.
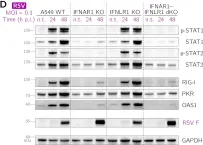
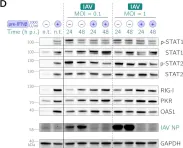
Type III interferons (IFN-λ1–λ4) are known to limit influenza virus infections in vivo and are non-redundant to type I interferons (IFN-α and IFN-β). Here, we demonstrated in vitro that type III interferons limit infection with influenza A virus (IAV) independently of STAT1 and STAT2 activation. Despite the fact that the knockout of the IFN-λ receptor (subunit IFNLR1), compared to the knockout of the IFN-β receptor (subunit IFNAR1), is associated with higher levels of STAT1/2 phosphorylation during infection, it results in a greater proportion of IAV-infected cells and higher viral RNA and protein levels. We showed that the ratio of dying to infected cells is lower in IFNLR1-deficient cells compared to wild-type cells suggesting that type III interferons limit the spread of IAV by promoting the death of IAV-infected cells. In contrast, type I interferons induce a stronger accumulation of proteins coded by interferon-stimulated genes, and correspondingly suppress IAV spread more effectively than type III interferons when provided prior to infection. Overall, our results suggest an additional non-transcriptional role of type III interferons in the control of viral infections. Key points IFNAR1 deficiency leads to a significant reduction of STAT1/2 activation in cell populations infected with IAV and a small decrease in IAV proliferation. IFNLR1 deficiency leads to a small reduction of STAT1/2 activation in cell populations infected with IAV and a significant increase in IAV proliferation. IFN-λ controls the proliferation of IAV (but not RSV) independently of STAT1/2 signaling. IFN-λ signaling increases the ratio of the dead to the infected cells, likely by promoting death in IAV-infected cells.
-
Homo sapiens (Human)
-
Immunology and Microbiology
Preprint on BioRxiv : the Preprint Server for Biology on 12 August 2024 by Preis, J. R., Rolvering, C., et al.
Resistance of melanoma cells to targeted therapy (BRAF and MEK inhibitors) is a major clinical problem and alternative treatments are sought. We describe the establishment of modular physiologic medium (MPM) and Mel-MPM (which contains additional supplements and sustains the 3D growth of melanoma cells, fibroblasts (NHDFs) and HMEC-1 endothelial cells) as novel resources for melanoma and combine them with a multi-cell-type matrix-embedded 3D culture model to investigate melanoma cell vulnerabilities in a more physiological setting. We made use of the modular nature of MPM to interrogate NEAA dependencies in melanoma cells and we found them to be particularly sensitive to the depletion of C/C. We additionally describe that melanoma cells are less sensitive to ferroptosis inducing compounds when cultured in MPM compared to RPMI and we could attribute this to different components of MPM and Mel-MPM (selenite, B27). Cell death induced by the glutathione peroxidase 4 inhibitor, ML162, had characteristics of ferroptosis or apoptosis depending on cell type, its drug resistance status and the culture medium. Cystine/cysteine starvation and ML162 treatment combinations increased melanoma cell death in 2D, 3D, and also in the complex matrix embedded multi-cell-type-3D system in Organoplates TM . This underlines the potential of combining metabolism-oriented drug treatments with amino acid starvation conditions, which is of interest in view of future therapeutic approaches to combat melanoma and other cancer types.
-
WB
-
Cancer Research
Antagonism between viral infection and innate immunity at the single-cell level.
In PLoS Pathogens on 1 September 2023 by Grabowski, F., Kochańczyk, M., et al.
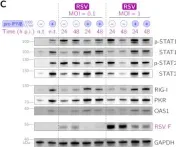
When infected with a virus, cells may secrete interferons (IFNs) that prompt nearby cells to prepare for upcoming infection. Reciprocally, viral proteins often interfere with IFN synthesis and IFN-induced signaling. We modeled the crosstalk between the propagating virus and the innate immune response using an agent-based stochastic approach. By analyzing immunofluorescence microscopy images we observed that the mutual antagonism between the respiratory syncytial virus (RSV) and infected A549 cells leads to dichotomous responses at the single-cell level and complex spatial patterns of cell signaling states. Our analysis indicates that RSV blocks innate responses at three levels: by inhibition of IRF3 activation, inhibition of IFN synthesis, and inhibition of STAT1/2 activation. In turn, proteins coded by IFN-stimulated (STAT1/2-activated) genes inhibit the synthesis of viral RNA and viral proteins. The striking consequence of these inhibitions is a lack of coincidence of viral proteins and IFN expression within single cells. The model enables investigation of the impact of immunostimulatory defective viral particles and signaling network perturbations that could potentially facilitate containment or clearance of the viral infection.
Copyright: © 2023 Grabowski et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
-
WB
-
Homo sapiens (Human)
-
Immunology and Microbiology
ADAR1 Biology Can Hinder Effective Antiviral RNA Interference.
In Journal of Virology on 27 April 2023 by Uhl, S., Jang, C., et al.
Viruses constantly evolve and adapt to the antiviral defenses of their hosts. The biology of viral circumvention of these selective pressures can often be attributed to the acquisition of novel antagonistic gene products or by rapid genome change that prevents host recognition. To study viral evasion of RNA interference (RNAi)-based defenses, we established a robust antiviral system in mammalian cells using recombinant Sendai virus designed to be targeted by endogenous host microRNAs (miRNAs) with perfect complementarity. Using this system, we previously demonstrated the intrinsic ability of positive-strand RNA viruses to escape this selective pressure via homologous recombination, which was not observed in negative-strand RNA viruses. Here, we show that given extensive time, escape of miRNA-targeted Sendai virus was enabled by host adenosine deaminase acting on RNA 1 (ADAR1). Independent of the viral transcript targeted, ADAR1 editing resulted in disruption of the miRNA-silencing motif, suggesting an intolerance for extensive RNA-RNA interactions necessary for antiviral RNAi. This was further supported in Nicotiana benthamiana, where exogenous expression of ADAR1 interfered with endogenous RNAi. Together, these results suggest that ADAR1 diminishes the effectiveness of RNAi and may explain why it is absent in species that utilize this antiviral defense system. IMPORTANCE All life at the cellular level has the capacity to induce an antiviral response. Here, we examine the result of imposing the antiviral response of one branch of life onto another and find evidence for conflict. To determine the consequences of eliciting an RNAi-like defense in mammals, we applied this pressure to a recombinant Sendai virus in cell culture. We find that ADAR1, a host gene involved in regulation of the mammalian response to virus, prevented RNAi-mediated silencing and subsequently allowed for viral replication. In addition, the expression of ADAR1 in Nicotiana benthamiana, which lacks ADARs and has an endogenous RNAi system, suppresses gene silencing. These data indicate that ADAR1 is disruptive to RNAi biology and provide insight into the evolutionary relationship between ADARs and antiviral defenses in eukaryotic life.
-
Genetics
-
Immunology and Microbiology
In J Virol on 23 November 2022 by Czerkies, M., Kochańczyk, M., et al.
Fig.1.D

-
WB
-
Collected and cropped from J Virol by CiteAb, provided under a CC-BY license
Image 1 of 11
In J Virol on 23 November 2022 by Czerkies, M., Kochańczyk, M., et al.
Fig.2.C

-
WB
-
Collected and cropped from J Virol by CiteAb, provided under a CC-BY license
Image 1 of 11
In J Virol on 23 November 2022 by Czerkies, M., Kochańczyk, M., et al.
Fig.2.D

-
WB
-
Collected and cropped from J Virol by CiteAb, provided under a CC-BY license
Image 1 of 11
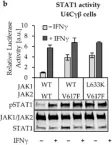
In Cancers (Basel) on 27 December 2019 by Raivola, J., Haikarainen, T., et al.
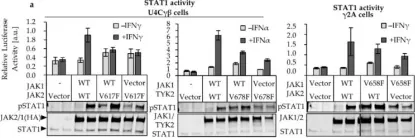
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 11
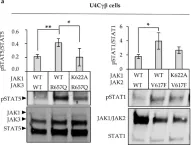
In Cancers (Basel) on 27 December 2019 by Raivola, J., Haikarainen, T., et al.
Fig.4.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 11
In Cancers (Basel) on 27 December 2019 by Raivola, J., Haikarainen, T., et al.
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 11
In Cancers (Basel) on 27 December 2019 by Raivola, J., Haikarainen, T., et al.
Fig.5.A

-
WB
-
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 11
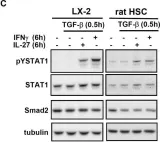
In Cell Commun Signal on 19 August 2010 by Schoenherr, C., Weiskirchen, R., et al.
Fig.5.C

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Cell Commun Signal by CiteAb, provided under a CC-BY license
Image 1 of 11
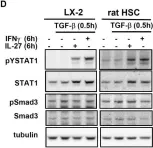
In Cell Commun Signal on 19 August 2010 by Schoenherr, C., Weiskirchen, R., et al.
Fig.5.D

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Cell Commun Signal by CiteAb, provided under a CC-BY license
Image 1 of 11
In PLoS Genet on 18 August 2006 by Chapgier, A., Boisson-Dupuis, S., et al.
Fig.4.A

-
WB
-
Collected and cropped from PLoS Genet by CiteAb, provided under a CC-BY license
Image 1 of 11
In PLoS Genet on 18 August 2006 by Chapgier, A., Boisson-Dupuis, S., et al.
Fig.7.C

-
WB
-
Collected and cropped from PLoS Genet by CiteAb, provided under a CC-BY license
Image 1 of 11