

Recently, we demonstrated that a novel bacterial cytotoxin, the protein MakA which is released by Vibrio cholerae, is a virulence factor, causing killing of Caenorhabditis elegans when the worms are grazing on the bacteria. Studies with mammalian cell cultures in vitro indicated that MakA could affect eukaryotic cell signalling pathways involved in lipid biosynthesis. MakA treatment of colon cancer cells in vitro caused inhibition of growth and loss of cell viability. These findings prompted us to investigate possible signalling pathways that could be targets of the MakA-mediated inhibition of tumour cell proliferation. Initial in vivo studies with MakA producing V. cholerae and C. elegans suggested that the MakA protein might target the PIP5K1α phospholipid-signalling pathway in the worms. Intriguingly, MakA was then found to inhibit the PIP5K1α lipid-signalling pathway in cancer cells, resulting in a decrease in PIP5K1α and pAkt expression. Further analyses revealed that MakA inhibited cyclin-dependent kinase 1 (CDK1) and induced p27 expression, resulting in G2/M cell cycle arrest. Moreover, MakA induced downregulation of Ki67 and cyclin D1, which led to inhibition of cell proliferation. This is the first report about a bacterial protein that may target signalling involving the cancer cell lipid modulator PIP5K1α in colon cancer cells, implying an anti-cancer effect.
© 2022. The Author(s).
Product Citations: 43
In Cell Death & Disease on 6 December 2022 by Toh, E., Baryalai, P., et al.
-
WB
-
Cancer Research
-
Cell Biology
In Scientific Reports on 17 July 2020 by Denu, R. A. & Burkard, M. E.
The centrosome is the microtubule organizing center of human cells and facilitates a myriad of cellular functions including organization of the mitotic spindle to ensure faithful chromosome segregation during mitosis, cell polarization and migration, and primary cilia formation. A numerical increase in centrosomes, or centrosome amplification (CA), is common in cancer and correlates with more aggressive clinical features and worse patient outcomes. However, the causes of CA in human cancer are unclear. Many previous studies have identified mechanisms of CA in cellulo, such as overexpression of PLK4, but it is unclear how often these are the primary mechanism in human disease. To identify a primary cause of CA, we analyzed The Cancer Genome Atlas (TCGA) genomic and transcriptomic data for genes encoding the 367 proteins that localize to the centrosome (the "centrosome-ome"). We identified the following candidates for primary causes of CA: gain-of-function alterations of CEP19, CEP72, CTNNB1, PTK2, NDRG1, SPATC1, TBCCD1; and loss-of-function alterations of CEP76, MCPH1, NEURL4, and NPM1. In cellulo analysis of these candidates revealed that loss of MCPH1/microcephalin caused the most robust increase in centriole number. MCPH1 deep gene deletions are seen in 5-15% of human cancers, depending on the anatomic site of the tumor. Mechanistic experiments demonstrated that loss of MCPH1 caused a CDK2-dependent increase in STIL levels at the centrosome to drive CA. We conclude that loss of MCPH1 is common in human cancer and is likely to be a cause of CA.
-
IF
-
Homo sapiens (Human)
-
Cancer Research
-
Cell Biology
Integrin-mediated adhesions in regulation of cellular senescence.
In Science Advances on 1 May 2020 by Shin, E. Y., Park, J. H., et al.
Bioinformatic and functional data link integrin-mediated cell adhesion to cellular senescence; however, the significance of and molecular mechanisms behind these connections are unknown. We now report that the focal adhesion-localized βPAK-interacting exchange factor (βPIX)-G protein-coupled receptor kinase interacting protein (GIT) complex controls cellular senescence in vitro and in vivo. βPIX and GIT levels decline with age. βPIX knockdown induces cellular senescence, which was prevented by reexpression. Loss of βPIX induced calpain cleavage of the endocytic adapter amphiphysin 1 to suppress clathrin-mediated endocytosis (CME); direct competition of GIT1/2 for the calpain-binding site on paxillin mediates this effect. Decreased CME and thus integrin endocytosis induced abnormal integrin signaling, with elevated reactive oxygen species production. Blocking integrin signaling inhibited senescence in human fibroblasts and mouse lungs in vivo. These results reveal a central role for integrin signaling in cellular senescence, potentially identifying a new therapeutic direction.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
In The Journal of Biological Chemistry on 9 March 2018 by Lee, J. G., Jung, E., et al.
Investigating stimulation of endogenous wound healing in corneal endothelial cells (CECs) may help address the global shortage of donor corneas by decreasing the number of transplants performed for blindness because of endothelial dysfunction. We previously reported that IL-1β stimulation leads to fibroblast growth factor (FGF2) expression, enhancing migration and proliferation of mammalian CECs. However, FGF2 also promotes the endothelial-mesenchymal transition, which can lead to retrocorneal membrane formation and blindness. This prompted us to investigate downstream FGF2 signaling targets that could be manipulated to prevent retrocorneal membrane formation. FGF2 stimulation altered cell morphology and induced expression of mesenchymal transition marker genes such as snail family transcriptional repressor 1 (SNAI1), SNAI2, zinc finger E-box-binding homeobox 1 (ZEB1), and ZEB2 This, in turn, induced expression of fibronectin, vimentin, and type I collagen, and suppressed E-cadherin in CECs in vitro and ex vivo siRNA-mediated SNAI1 knockdown revealed that SNAI1 induces ZEB1 expression, in turn inducing expression of type I collagen, the major component of retrocorneal membranes, and of cyclin-dependent kinase 2 (CDK2) and cyclin E1, promoting cell proliferation. siRNA-mediated knockdown of SNAI1 or ZEB1, but not of CDK2, inhibited FGF2-dependent expression of fibronectin, vimentin, and type I collagen and of suppression of E-cadherin expression. We conclude that SNAI1 is a key regulator of FGF2-dependent mesenchymal transition in human ex vivo corneal endothelium, with ZEB1 regulating type I collagen expression and CDK2 regulating cell proliferation. These results suggest that SNAI1 promotes fibrosis and cell proliferation in human corneal endothelium through ZEB1 and CDK2.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
Drp1 regulates mitochondrial morphology and cell proliferation in cutaneous squamous cell carcinoma.
In Journal of Dermatological Science on 1 December 2017 by Kitamura, S., Yanagi, T., et al.
Dynamin-related protein 1 (Drp1) mediates mitochondrial fission. Recently, several studies have shown that Drp1 plays an important role in some cancers. However, little is known about Drp1 in cutaneous squamous cell carcinoma (SCC).
To investigate the role of Drp1 in the tumorigenesis of cutaneous SCCs.
We investigated cell proliferation, cell cycle, mitochondrial morphology, and MAPK signaling pathway using cutaneous SCC A431 and DJM1 cells that were transfected with shRNA vectors targeting Drp1. The Drp1 gene-knockdown SCC cells showed lower cell proliferation than scramble-control cells, as assessed by direct cell counting and clonogenic assays. DNA content analysis showed Drp1 knockdown to cause G2/M arrest. Morphologically, the depletion of Drp1 resulted in an elongated, hyper-fused mitochondrial network. The MEK inhibitor PD325901 suppressed cell proliferation, as well as inhibiting the phosphorylation of ERK1/2 and Drp1Ser616. Also, PD325901 caused the dysregulation of the mitochondrial network. In tumor xenografts of DJM1 cells, the knockdown of Drp1 suppressed tumor growth in vivo, and clinically, the expression levels of Drp1 were higher in cutaneous SCCs than in normal epidermis, and correlated positively with the advanced clinical stages.
Our results reveal a crucial function for Drp1 in regulating tumor growth, mitochondrial morphology, and cell cycle in cutaneous SCC, suggesting that Drp1 could be a novel target for skin tumor therapies.
Copyright © 2017 Japanese Society for Investigative Dermatology. Published by Elsevier B.V. All rights reserved.
-
Cancer Research
-
Cell Biology
In Cell Death Dis on 6 December 2022 by Toh, E., Baryalai, P., et al.
Fig.4.A

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 8
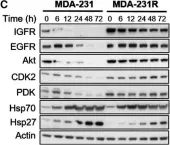
In Mol Oncol on 1 May 2017 by Chai, R. C., Vieusseux, J. L., et al.
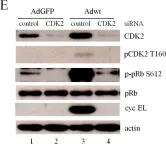
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Mol Oncol by CiteAb, provided under a CC-BY license
Image 1 of 8
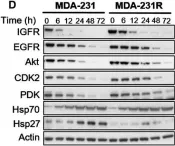
In Mol Oncol on 1 May 2017 by Chai, R. C., Vieusseux, J. L., et al.
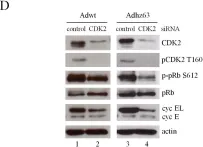
Fig.4.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Mol Oncol by CiteAb, provided under a CC-BY license
Image 1 of 8
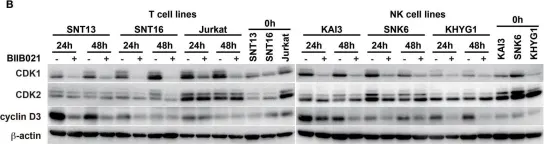
In Front Microbiol on 29 April 2015 by Suzuki, M., Takeda, T., et al.
Fig.5.B

-
WB
-
Collected and cropped from Front Microbiol by CiteAb, provided under a CC-BY license
Image 1 of 8
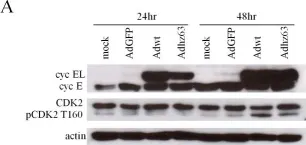
In PLoS One on 26 February 2013 by Cheng, P. H., Rao, X. M., et al.
Fig.3.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 8
In PLoS One on 26 February 2013 by Cheng, P. H., Rao, X. M., et al.
Fig.7.D

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 8
In PLoS One on 26 February 2013 by Cheng, P. H., Rao, X. M., et al.
Fig.8.E

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 8
In BMC Cancer on 25 August 2011 by Wedel, S., Hudak, L., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from BMC Cancer by CiteAb, provided under a CC-BY license
Image 1 of 8