


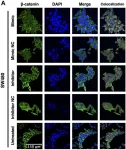

Wnt signaling plays a pivotal role during development and homeostasis. Upon pathway activation, CTNNB1 (also known as beta-catenin) drives the expression of target genes from regulatory regions bound by TCF/LEF transcription factors. Gene regulation, however, entails the interplay between sequence information and 3D genome structure, yet the impact of Wnt signaling on genome structure has been poorly explored. Here, we investigate how Wnt signaling influences CTCF and cohesin, key regulators of 3D genome organization. We identify a series of novel CTCF binding sites that emerge upon Wnt stimulation: CTCF Redistributions Under Wnt (RUW). RUW sites are characterized by CTCF, cohesin, and TCF/LEF occupancy, and are dependent on beta-catenin. Beta-catenin and CTCF colocalize upon pathway activation, and disruption of selected binding sites perturbs target gene regulation. Moreover, Wnt signaling reorganizes the 3D genome as evidenced by genome-wide alterations in CTCF-bound loops. This work reveals a previously unexplored role for CTCF in the regulation of Wnt signaling.
© 2025 Nordin et al.; Published by Cold Spring Harbor Laboratory Press.
Product Citations: 1,259
In Genome Research on 14 July 2025 by Nordin, A., Chakraborty, C., et al.
A multicellular star-shaped actin network underpins epithelial organization and connectivity.
In Nature Communications on 4 July 2025 by Barai, A., Soleilhac, M., et al.
Epithelial tissues withstand external stresses while maintaining structural stability. Bicellular junctions and the actomyosin network support epithelial integrity, packing and remodelling. While their role in development and disease are well studied, their synergistic impact on maintaining tissue organization remains unclear. Here, we identify a tissue-scale actomyosin network in adult murine intestinal villi, as well as in an ex vivo organoid-based epithelium model. This actomyosin network consists of repeated units of actin stars - radial actin structures at the base of hexagonal cells - linked via bicellular junctions into a multicellular array. Functionally, actin stars maintain epithelial morphological stability by preserving cell shape and packing. Laser ablation experiments support a modified vertex model, linking tension along actin star branches to epithelial arrangement. Additionally, actin stars act as basal locks, limiting protrusive activity, and hindering cell migration and tissue disruption. Together, these findings reveal the star-shaped supracellular actin network as a pivotal biomechanical system governing epithelial layer coordination.
© 2025. The Author(s).
-
Cell Biology
In Scientific Reports on 4 July 2025 by Lei, Y., Hu, H., et al.
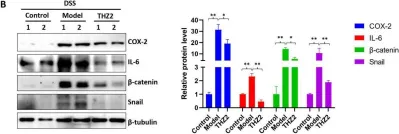
Hyperhomocysteinemia can cause severe damage to kidney. Ferroptosis represents a critical mechanism in the initiation and development of kidney disorders. We focus on the β-catenin/GPX4 signaling pathway to explore how homocysteine influences ferroptosis regulation in renal tubular epithelial cells. C57BL/6J mice were administered drinking water with high level of homocysteine to establish a hyperhomocysteinemia model. In the cell experiments, HKC-8 cells were exposed to homocysteine for a duration of 12 h. Active β-catenin, β-catenin, GPX4, FTH1, and KIM-1 were detected using Western blotting; Biochemical assays were conducted to measure lipid ROS, Fe2+, and GSH; GPX4 and β-catenin were detected through immunohistochemistry and immunofluorescence techniques; Mitochondrial damage was examined using transmission electron microscopy; ChIP analysis, coupled with dual-luciferase reporter gene assays, was employed to investigate the relationship between β-catenin protein and GPX4 gene promoter. Our findings revealed that homocysteine disrupted β-catenin signaling, inhibited GPX4 expression in renal tubular epithelial cells, subsequently promoted ferroptosis. Overexpression of β-catenin or GPX4 inhibited ferroptosis induced by homocysteine, and β-catenin regulated GPX4 expression in renal tubular epithelial cells. Further assays demonstrated that GPX4 acted as a target gene of β-catenin. In conclusion, homocysteine elicits ferroptosis in renal tubular epithelial cells by disrupting β-catenin signaling and inhibiting its target gene, GPX4.
© 2025. The Author(s).
-
WB
-
IHC
In Scientific Reports on 1 July 2025 by Lei, Y., Hu, H., et al.
Homocysteine can cause damage to cardiomyocytes. However, Mitophagy is essential for preserving homeostasis in cardiomyocytes. So, we focused on investigating the impact of homocysteine on cardiomyocyte mitophagy and cardiac hypertrophy through the β-catenin/FUNDC1 pathway. Mice were administered water containing homocysteine (1.8 g/L) to induce hyperhomocysteinemia for 4 weeks. The overexpression of specific genes, including β-catenin and FUNDC1, were performed by gene delivery mediated with adeno-associated virus. In vitro, cardiomyocytes were exposed to homocysteine (1 mmol/L) and then transfected with plasmids to overexpress β-catenin and FUNDC1, respectively. The duration of cell experiments was 48 h. Western blotting was employed to assess the expression levels of β-catenin, active β-catenin, FUNDC1, LC3, p62, α-actin, and β-MHC. Immunohistochemistry and immunofluorescence techniques were applied to measure β-catenin and FUNDC1 in cardiomyocytes. Cell viability was assessed using a CCK-8 assay kit, and mitophagy was observed under transmission electron microscopy. The interaction between β-catenin protein and the promoter of the FUNDC1 gene was examined using ChIP assay and dual-luciferase reporter gene assay. Homocysteine inhibited β-catenin signaling and the FUNDC1-mediated mitophagy in the cardiomyocytes, simultaneously promoting cardiac hypertrophy in vitro and in vivo. Elevated β-catenin signaling promoted FUNDC1 expression, then restored the normal level of mitophagy, and consequently inhibited homocysteine-induced cardiac hypertrophy. Similarly, overexpression of FUNDC1 restored mitophagy and protected cardiomyocytes from hypertrophy. In addition, FUNDC1 served as a target gene of β-catenin. In summary, homocysteine induces cardiomyocyte hypertrophy by inhibiting β-catenin signaling and suppressing FUNDC1-mediated mitophagy.
© 2025. The Author(s).
-
WB
-
IHC
-
Mus musculus (House mouse)
In Frontiers in Pharmacology on 9 June 2025 by Lei, Y., Xu, L., et al.
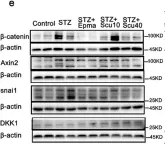
The chemotherapeutic agent doxorubicin has the side effect of inducing injury to cardiomyocytes. Ferroptosis plays an essential role in the onset and progression of cardiac injury. Ophiopogonin D is considered the active component of the Chinese herbal medicine Mai Dong, which is commonly used for the treatment of cardiovascular diseases. This study investigates the impact of ophiopogonin D on doxorubicin-induced cardiomyocyte ferroptosis by focusing on the β-catenin/GPX4 signaling pathway.
Mice were injected intraperitoneally with doxorubicin (10 mg/kg) to create a model of cardiotoxicity. Cardiomyocytes exposed to doxorubicin (1 μM) were treated with ophiopogonin D (5 μM). Western blotting was used to detect β-catenin, FTH1, and GPX4. Malondialdehyde (MDA), glutathione (GSH), and Fe2+ levels were measured using biochemical assays. In addition, GPX4 expression was detected by immunohistochemistry and immunofluorescence staining. Mitochondrial injury was examined by transmission electron microscopy. Chromatin immunoprecipitation (ChIP) combined with dual-luciferase reporter gene assay was used to analyze the interaction between β-catenin and the promoter of the GPX4 gene.
Doxorubicin inhibited β-catenin activity and GPX4 expression, promoting cardiomyocyte ferroptosis in vitro and in vivo. Ophiopogonin D increased β-catenin expression and promoted GPX4 expression, thereby inhibiting doxorubicin-induced ferroptosis in cardiomyocytes. Moreover, β-catenin overexpression enhanced GPX4 expression and alleviated homocysteine-induced ferroptosis in cardiomyocytes. Furthermore, results from the ChIP and dual-luciferase reporter assays indicated that GPX4 acted as a target gene of β-catenin.
Ophiopogonin D inhibits cardiomyocyte ferroptosis induced by doxorubicin by restoring the β-catenin/GPX4 signaling pathway.
Copyright © 2025 Lei, Xu, Liu and Zhao.
-
WB
-
IHC
-
Pharmacology
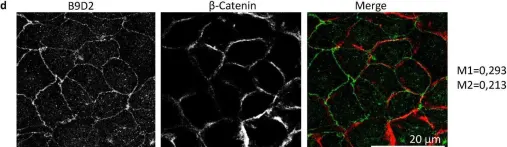
In Sci Rep on 25 October 2024 by Caenen-Braz, C., Bouzhir, L., et al.
Fig.2.D

-
IHC-IF
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 273
In Sci Rep on 24 August 2024 by Nilkhet, S., Vongthip, W., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 273
In Sci Rep on 24 August 2024 by Nilkhet, S., Vongthip, W., et al.
Fig.4.C

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 273
In Sci Rep on 24 August 2024 by Nilkhet, S., Vongthip, W., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 273
In Sci Rep on 24 August 2024 by Nilkhet, S., Vongthip, W., et al.
Fig.2.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 273
In Front Cell Dev Biol on 23 July 2024 by Lechuga, S., Marino-Melendez, A., et al.
Fig.3.B

-
ICC-IF
-
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In Front Pharmacol on 25 April 2024 by Escalante, P. I., Quiñones, L. A., et al.
Fig.3.B

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from Front Pharmacol by CiteAb, provided under a CC-BY license
Image 1 of 273
In Front Pharmacol on 25 April 2024 by Escalante, P. I., Quiñones, L. A., et al.
Fig.3.A

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from Front Pharmacol by CiteAb, provided under a CC-BY license
Image 1 of 273
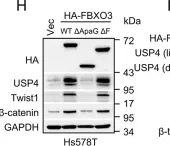
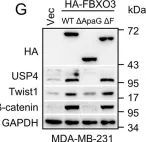
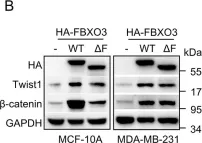
In Nat Prod Bioprospect on 24 April 2024 by Huang, B., Han, R., et al.
Fig.4.E

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Prod Bioprospect by CiteAb, provided under a CC-BY license
Image 1 of 273
In Biomedicines on 18 March 2024 by Wang, S. T., Wang, Y. Y., et al.
Fig.2.A

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from Biomedicines by CiteAb, provided under a CC-BY license
Image 1 of 273
In Biomedicines on 18 March 2024 by Wang, S. T., Wang, Y. Y., et al.
Fig.2.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Biomedicines by CiteAb, provided under a CC-BY license
Image 1 of 273
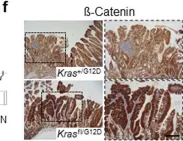
In Nat Commun on 2 January 2024 by Najumudeen, A. K., Fey, S. K., et al.
Fig.5.F

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 273
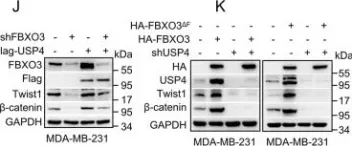
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.3.F

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.4.E

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.5.B

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.6.A

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.3.J

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.3.H

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.3.G

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
In PLoS Biol on 1 December 2023 by Xu, J., Guo, R., et al.
Fig.2.B

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 273
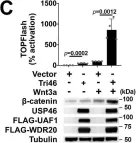
In Nat Commun on 5 October 2023 by Ng, V. H., Spencer, Z., et al.
Fig.1.C

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 273
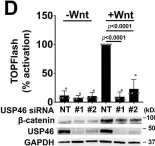
In Nat Commun on 5 October 2023 by Ng, V. H., Spencer, Z., et al.
Fig.1.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 273
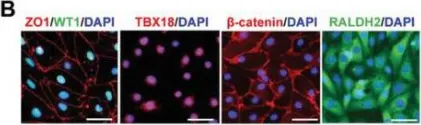
In Adv Sci (Weinh) on 1 September 2023 by Luo, X. L., Jiang, Y., et al.
Fig.1.B

-
ICC-IF
-
Collected and cropped from Adv Sci (Weinh) by CiteAb, provided under a CC-BY license
Image 1 of 273
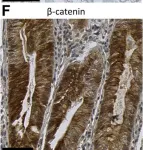
In Dis Model Mech on 1 August 2023 by Cerretelli, G., Zhou, Y., et al.
Fig.2.E

-
IHC
-
Collected and cropped from Dis Model Mech by CiteAb, provided under a CC-BY license
Image 1 of 273
In Biol Open on 15 May 2023 by Yoshimoto, S., Taguchi, M., et al.
Fig.1.C

-
IHC
-
Collected and cropped from Biol Open by CiteAb, provided under a CC-BY license
Image 1 of 273