
Reactivation of p53 tumor-suppressor function by small molecules is an attractive strategy to defeat cancer. A potent p53-reactivating molecule RITA, which triggers p53-dependent apoptosis in human tumor cells in vitro and in vivo, exhibits p53-independent cytotoxicity due to modifications by detoxification enzyme Sulfotransferase 1A1 (SULT1A1), producing a reactive carbocation. Several synthetic modifications to RITA's heterocyclic scaffold lead to higher energy barriers for carbocation formation. In this study, we addressed the question whether RITA analogs NSC777196 and NSC782846 can induce p53-dependent apoptosis without SULT1A1-dependent DNA damage. We found that RITA analog NSC782846, but not NSC777196, induced p53-regulated genes, targeted oncogene addiction, and killed cancer cells upon p53 reactivation, but without induction of DNA damage and inhibition RNA pol II. Our results might demonstrate a method for designing more specific and potent RITA analogs to accelerate translation of p53-targeting compounds from laboratory bench to clinic.
©2022 The Authors; Published by the American Association for Cancer Research.
Product Citations: 47
Decreased DNA Damage and Improved p53 Specificity of RITA Analogs.
In Molecular Cancer Therapeutics on 7 October 2022 by Zhan, Y., Zhou, X., et al.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
-
Genetics
Enterobacteria impair host p53 tumor suppressor activity through mRNA destabilization.
In Oncogene on 1 April 2022 by Aschtgen, M. S., Fragkoulis, K., et al.
Increasing evidence highlights the role of bacteria in the physiopathology of cancer. However, the underlying molecular mechanisms remains poorly understood. Several cancer-associated bacteria have been shown to produce toxins which interfere with the host defense against tumorigenesis. Here, we show that lipopolysaccharides from Klebsiella pneumoniae and other Enterobacteria strongly inhibit the host tumor suppressor p53 pathway through a novel mechanism of p53 regulation. We found that lipopolysaccharides destabilize TP53 mRNA through a TLR4-NF-κB-mediated inhibition of the RNA-binding factor Wig-1. Importantly, we show that K. pneumoniae disables two major tumor barriers, oncogene-induced DNA damage signaling and senescence, by impairing p53 transcriptional activity upon DNA damage and oncogenic stress. Furthermore, we found an inverse correlation between the levels of TLR4 and p53 mutation in colorectal tumors. Hence, our data suggest that the repression of p53 by Enterobacteria via TLR4 alleviates the selection pressure for p53 oncogenic mutations and shapes the genomic evolution of cancer.
© 2022. The Author(s).
-
WB
-
Cancer Research
-
Genetics
Temporal proteomics reveal specific cell cycle oncoprotein downregulation by p97/VCP inhibition.
In Cell Chemical Biology on 17 March 2022 by Wang, F., Li, S., et al.
Targeting protein quality control (PQC) pathways using proteasome or p97/VCP inhibition can effectively treat blood tumors. However, in solid tumors, only p97/VCP inhibitors are effective. To probe this difference in efficacy, we tracked HCT116 colon cancer cells using temporal proteomics to define the cellular and molecular responses to proteasome and p97 inhibition. Proteins involved in general PQC pathways were similarly upregulated by both treatments, suggesting that the proteotoxic stress caused by inhibitors does not explain the differential therapeutic effectiveness. Unexpectedly, proteins specifically dysregulated by two p97 inhibitors are involved in cell cycle control. Indeed, eleven cell cycle proteins were downregulated by p97 inhibition but not by proteasome inhibition. Western blot analysis validated the degradation of cyclin D1 and Securin, which depends on proteasome but not on p97. Differing regulation of cell cycle proteins by p97 and the proteasome may, therefore, explain the therapeutic efficacy of p97 inhibitors in colon cancer.
Copyright © 2021 Elsevier Ltd. All rights reserved.
In Cancers on 17 March 2021 by Ungefroren, H., Christl, J., et al.
Autocrine transforming growth factor β (aTGFβ) has been implicated in the regulation of cell invasion and growth of several malignant cancers such as pancreatic ductal adenocarcinoma (PDAC) or triple-negative breast cancer (TNBC). Recently, we observed that endogenous TGFB1 can inhibit rather than stimulate cell motility in cell lines with high aTGFβ production and mutant KRAS, i.e., Panc1 (PDAC) and MDA-MB-231 (TNBC). The unexpected anti-migratory role prompted us to evaluate if aTGFβ1 may be able to antagonize the action of exogenous (recombinant human) TGFβ (rhTGFβ), a well-known promoter of cell motility and growth arrest in these cells. Surprisingly, RNA interference-mediated knockdown of the endogenous TGFB1 sensitized genes involved in EMT and cell motility (i.e., SNAI1) to up-regulation by rhTGFβ1, which was associated with a more pronounced migratory response following rhTGFβ1 treatment. Ectopic expression of TGFB1 decreased both basal and rhTGFβ1-induced migratory activities in MDA-MB-231 cells but had the opposite effect in Panc1 cells. Moreover, silencing TGFB1 reduced basal proliferation and enhanced growth inhibition by rhTGFβ1 and induction of cyclin-dependent kinase inhibitor, p21WAF1. Finally, we show that aTGFβ1 promotes MEK-ERK signaling and vice versa to form a self-perpetuating feedforward loop that is sensitive to SB431542, an inhibitor of the TGFβ type I receptor, ALK5. Together, these data suggest that in transformed cells an ALK5-MEK-ERK-aTGFβ1 pathway opposes the promigratory and growth-arresting function of rhTGFβ1. This observation has profound translational implications for TGFβ signaling in cancer.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
-
Endocrinology and Physiology
In Proceedings of the National Academy of Sciences of the United States of America on 2 June 2020 by Li, Y. C., Lytle, N. K., et al.
HRAS, NRAS, and KRAS4A/KRAS4B comprise the RAS family of small GTPases that regulate signaling pathways controlling cell proliferation, differentiation, and survival. RAS pathway abnormalities cause developmental disorders and cancers. We found that KRAS4B colocalizes on the cell membrane with other RAS isoforms and a subset of prenylated small GTPase family members using a live-cell quantitative split luciferase complementation assay. RAS protein coclustering is mainly mediated by membrane association-facilitated interactions (MAFIs). Using the RAS-RBD (CRAF RAS binding domain) interaction as a model system, we showed that MAFI alone is not sufficient to induce RBD-mediated RAS inhibition. Surprisingly, we discovered that high-affinity membrane-targeted RAS binding proteins inhibit RAS activity and deplete RAS proteins through an autophagosome-lysosome-mediated degradation pathway. Our results provide a mechanism for regulating RAS activity and protein levels, a more detailed understanding of which should lead to therapeutic strategies for inhibiting and depleting oncogenic RAS proteins.
-
Homo sapiens (Human)
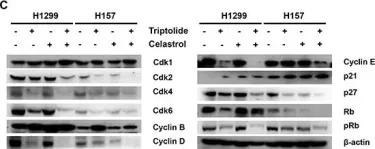
In J Cell Mol Med on 1 September 2018 by Tian, Y., Chen, H., et al.
Fig.1.B

-
WB
-
Collected and cropped from J Cell Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 5
In Oncotarget on 20 October 2015 by Jiang, Q. W., Cheng, K. J., et al.
Fig.3.C

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 5
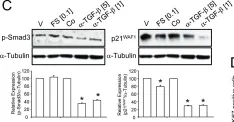
In PLoS One on 24 February 2015 by Ungefroren, H., Hyder, A., et al.
Fig.5.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5
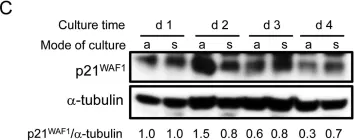
In PLoS One on 24 February 2015 by Ungefroren, H., Hyder, A., et al.
Fig.1.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5
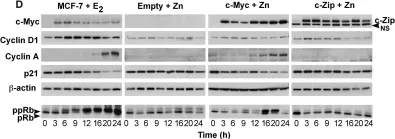
In PLoS One on 21 August 2008 by Musgrove, E. A., Sergio, C. M., et al.
Fig.1.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5