


Identifying effective therapies targeting multi-protein complexes that lack catalytic sites or cofactor pockets remains a long-standing challenge. The proto-oncogene, ubiquitin E3 ligase SCFSkp2, is one such target. SCFSkp2 promotes the proteasomal degradation of the cyclin-dependent kinase inhibitor p27, which controls cell cycle progression. Targeted knockout of Rb1/Trp53 causes metastatic prostate cancer in mice; additional knockout of Skp2 completely blocks tumorigenesis. We compared gene-edited mice that carried two different single amino acid changes in the SCFSkp2 complex, structurally predicted to inhibit the degradation of p27. Mutation of the SCFSkp2 accessory protein Cks1 (Cks1N45R) completely blocked Rb1/Trp53-driven prostate tumorigenesis, phenocopying Skp2 knockout, whereas a mutation directly stabilizing p27 (p27T187A) did not. This was consistent with structural models that predicted the binding of both p27 and p27T187A to the SCFSkp2/Cks1/Cdk2/CyclinA/p27 complex, and their subsequent ubiquitination and degradation, albeit at different rates. Two binding modes, which differ in their dependence on phosphorylated T187, are predicted by the model. Studies confirmed the role of p27 in mediating tumorigenesis in Rb1/Trp53 mutant tumors and revealed a mutually destabilizing Skp2 and p27 feedback loop. The integration of gene editing, drug-surrogate mutations, and mouse tumor models offers a blueprint for studying SCFSkp2 and other multi-subunit biomedical targets.
© 2025. The Author(s).
Product Citations: 145
In Communications Biology on 22 February 2025 by Xue, Y., Zhu, L., et al.
In Signal Transduction and Targeted Therapy on 20 January 2025 by Kim, J. E., Kim, H. S., et al.
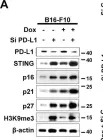
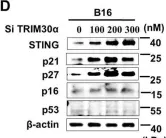
Dynamic communication between hepatocytes and the environment is critical in hepatocellular carcinoma (HCC) development. Clinical immunotherapy against HCC is currently unsatisfactory and needs more systemic considerations, including the identification of new biomarkers and immune checkpoints. Transmembrane 4 L six family member 5 (TM4SF5) is known to promote HCC, but it remains unclear how cancerous hepatocytes avoid immune surveillance and whether avoidance can be blocked. We investigated how TM4SF5-mediated hepatic tumorigenesis avoids surveillance by natural killer (NK) cells, which are prevalent in the liver, and whether the avoidance can be blocked by anti-TM4SF5 agents. We used comprehensive structure activity relationship analysis to identify TM4SF5-specific isoxazole (TSI)-based small molecules that inhibit TM4SF5-mediated effects. TM4SF5 expressed by hepatocytes reduced NK cell cytotoxicity by downregulating stimulatory ligands/receptors, including signaling lymphocytic activation molecule family member 7 (SLAMF7). TM4SF5 bound SLAMF7 depending on N-glycosylation and caused intracellular trafficking of SLAMF7 from the plasma membrane to lysosomes for degradation. TSI treatments in cell lines and animal models of HCC blocked this binding, intracellular trafficking, and downregulation, resulting in higher levels of stimulatory NK cell ligands. In mouse xenograft models, TSI treatment abrogated HCC development by increasing the abundance and dispersion of Slamf7-positive cells in liver tissues, recapitulating the phenotype of Tm4sf5-knockout mice and indicating TSI-mediated restoration of NK cell surveillance. These findings suggest that TSIs can inhibit TM4SF5-mediated liver carcinogenesis by increasing NK cell surveillance.
© 2025. The Author(s).
-
WB
-
Immunology and Microbiology
Effects of phospholipase D1-inhibitory peptide on the growth and metastasis of gastric cancer cells.
In Molecules and Cells on 1 November 2024 by Kim, D., Yoon, M. S., et al.
Phospholipase D1 (PLD1) contributes to cancer development and progression through its effects on cell proliferation, survival, invasion, metastasis, angiogenesis, drug resistance, and modulation of the tumor microenvironment. Its central role in these processes makes it a promising target for novel cancer treatments aimed at inhibiting its activity and disrupting the signaling pathways it regulates. In this study, we aimed to investigate the effect of PLD1 inhibition on gastric cancer cell growth using a novel peptide inhibitor, TAT-TVTSP. PLD1, which plays a role in cancer progression, catalyzes the conversion of phosphatidylcholine into choline and phosphatidic acid through hydrolysis. To effectively target PLD1 in cells, we engineered TAT-TVTSP by fusing a PLD1-inhibitory peptide (TVTSP) with a cell-penetrating peptide (TAT). We observed that TAT-TVTSP effectively inhibited PLD1 activity in AGS gastric cancer cells. Moreover, TAT-TVTSP significantly inhibited the mammalian target of the rapamycin signaling pathway, including the phosphorylation of key downstream targets such as S6K1, AKT, S473, glycogen synthase kinase-3b, and forkhead box O1. TAT-TVTSP did not induce cell death, but it triggered cell cycle arrest by activating p21 and p27 via AKT phosphorylation. Functional assays revealed that TAT-TVTSP significantly impaired the colony-forming ability of AGS cells, thus inhibiting cell proliferation. Transwell and wound-healing assays revealed that this peptide disrupted the cellular behaviors critical to cancer progression, such as migration and invasion. In vivo, TAT-TVTSP significantly reduced tumor growth in the xenograft model of gastric cancer without any toxicity. Overall, our results suggest that TAT-TVTSP is a novel therapeutic agent for PLD1-mediated cancers.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
-
Cancer Research
PPTC7 antagonizes mitophagy by promoting BNIP3 and NIX degradation via SCFFBXL4.
In EMBO Reports on 1 August 2024 by Nguyen, G. D. T., Townsend, B., et al.
Mitophagy must be carefully regulated to ensure that cells maintain appropriate numbers of functional mitochondria. The SCFFBXL4 ubiquitin ligase complex suppresses mitophagy by controlling the degradation of BNIP3 and NIX mitophagy receptors, and FBXL4 mutations result in mitochondrial disease as a consequence of elevated mitophagy. Here, we reveal that the mitochondrial phosphatase PPTC7 is an essential cofactor for SCFFBXL4-mediated destruction of BNIP3 and NIX, suppressing both steady-state and induced mitophagy. Disruption of the phosphatase activity of PPTC7 does not influence BNIP3 and NIX turnover. Rather, a pool of PPTC7 on the mitochondrial outer membrane acts as an adaptor linking BNIP3 and NIX to FBXL4, facilitating the turnover of these mitophagy receptors. PPTC7 accumulates on the outer mitochondrial membrane in response to mitophagy induction or the absence of FBXL4, suggesting a homoeostatic feedback mechanism that attenuates high levels of mitophagy. We mapped critical residues required for PPTC7-BNIP3/NIX and PPTC7-FBXL4 interactions and their disruption interferes with both BNIP3/NIX degradation and mitophagy suppression. Collectively, these findings delineate a complex regulatory mechanism that restricts BNIP3/NIX-induced mitophagy.
© 2024. The Author(s).
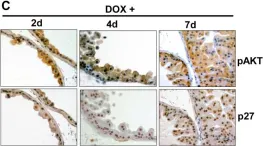
In Oncogene on 1 March 2024 by Wang, J., Ferrena, A., et al.
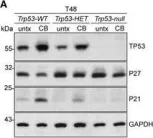
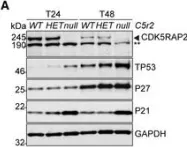
Osteosarcoma(OS) is a highly aggressive bone cancer for which treatment has remained essentially unchanged for decades. Although OS is characterized by extensive genomic heterogeneity and instability, RB1 and TP53 have been shown to be the most commonly inactivated tumor suppressors in OS. We previously generated a mouse model with a double knockout (DKO) of Rb1 and Trp53 within cells of the osteoblastic lineage, which largely recapitulates human OS with nearly complete penetrance. SKP2 is a repression target of pRb and serves as a substrate recruiting subunit of the SCFSKP2 complex. In addition, SKP2 plays a central role in regulating the cell cycle by ubiquitinating and promoting the degradation of p27. We previously reported the DKOAA transgenic model, which harbored a knock-in mutation in p27 that impaired its binding to SKP2. Here, we generated a novel p53-Rb1-SKP2 triple-knockout model (TKO) to examine SKP2 function and its potential as a therapeutic target in OS. First, we observed that OS tumorigenesis was significantly delayed in TKO mice and their overall survival was markedly improved. In addition, the loss of SKP2 also promoted an apoptotic microenvironment and reduced the stemness of DKO tumors. Furthermore, we found that small-molecule inhibitors of SKP2 exhibited anti-tumor activities in vivo and in OS organoids as well as synergistic effects when combined with a standard chemotherapeutic agent. Taken together, our results suggest that SKP2 inhibitors may reduce the stemness plasticity of OS and should be leveraged as next-generation adjuvants in this cancer.
© 2024. The Author(s).
-
Cancer Research
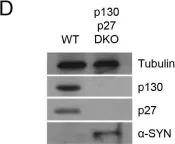
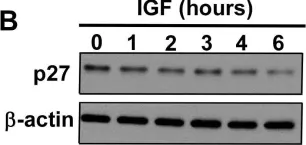
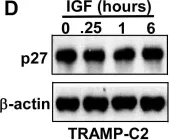
In Signal Transduct Target Ther on 20 January 2025 by Kim, J. E., Kim, H. S., et al.
Fig.2.D

-
WB
-
Collected and cropped from Signal Transduct Target Ther by CiteAb, provided under a CC-BY license
Image 1 of 25
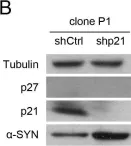
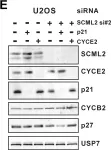
In Cell Death Dis on 15 September 2022 by Lee, J. J., Kim, S. Y., et al.
Fig.3.A

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 25
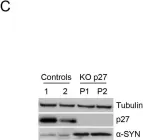
In EMBO J on 18 July 2022 by Tátrai, P. & Gergely, F.
Fig.6.A

-
WB
-
Collected and cropped from EMBO J by CiteAb, provided under a CC-BY license
Image 1 of 25
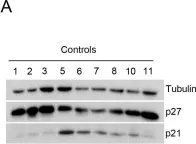
In EMBO J on 18 July 2022 by Tátrai, P. & Gergely, F.
Fig.7.A

-
WB
-
Collected and cropped from EMBO J by CiteAb, provided under a CC-BY license
Image 1 of 25
In Cell Death Discov on 8 February 2021 by Lee, J. J., Park, I. H., et al.
Fig.3.D

-
WB
-
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 27 March 2018 by Gallastegui, E., Domuro, C., et al.
Fig.3.D

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 27 March 2018 by Gallastegui, E., Domuro, C., et al.
Fig.2.B

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 27 March 2018 by Gallastegui, E., Domuro, C., et al.
Fig.1.C

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 27 March 2018 by Gallastegui, E., Domuro, C., et al.
Fig.1.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 27 March 2018 by Gallastegui, E., Domuro, C., et al.
Fig.1.E

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 2 March 2018 by Nakamura, A., Nakata, D., et al.
Fig.3.B

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Sci Rep on 16 May 2016 by Ferrán, B., Martí-Pàmies, I., et al.
Fig.7.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 25
In Sci Rep on 16 May 2016 by Ferrán, B., Martí-Pàmies, I., et al.
Fig.5.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 20 February 2015 by Vosper, J., Masuccio, A., et al.
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In Oncotarget on 20 February 2015 by Vosper, J., Masuccio, A., et al.
Fig.3.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 25
In J Cardiothorac Surg on 4 February 2014 by Redondo, S., Navarro-Dorado, J., et al.
Fig.2.A

-
WB
-
Collected and cropped from J Cardiothorac Surg by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS Biol on 1 December 2013 by Lecona, E., Rojas, L. A., et al.
Fig.7.A

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS Biol on 1 December 2013 by Lecona, E., Rojas, L. A., et al.
Fig.5.A

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS Biol on 1 December 2013 by Lecona, E., Rojas, L. A., et al.
Fig.4.E

-
WB
-
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS Biol on 1 December 2013 by Lecona, E., Rojas, L. A., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS One on 31 July 2012 by Wang, H., Xu, Y., et al.
Fig.5.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS One on 31 July 2012 by Wang, H., Xu, Y., et al.
Fig.5.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS One on 31 July 2012 by Wang, H., Xu, Y., et al.
Fig.4.C

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS One on 31 July 2012 by Wang, H., Xu, Y., et al.
Fig.4.B

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 25
In PLoS Biol on 1 December 2003 by Trotman, L. C., Niki, M., et al.
Fig.5.C

-
IHC
-
Mus musculus (House mouse)
Collected and cropped from PLoS Biol by CiteAb, provided under a CC-BY license
Image 1 of 25