

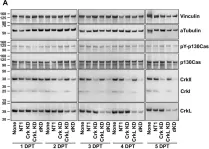

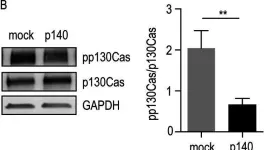
Acinar to ductal metaplasia is the prerequisite for the initiation of Kras-driven pancreatic ductal adenocarcinoma (PDAC), and candidate genes regulating this process are emerging from genome-wide association studies. The adaptor protein p130Cas emerged as a potential PDAC susceptibility gene and a Kras-synthetic lethal interactor in pancreatic cell lines; however, its role in PDAC development has remained largely unknown.Human PDAC samples and murine KrasG12D-dependent pancreatic cancer models of increasing aggressiveness were used. p130Cas was conditionally ablated in pancreatic cancer models to investigate its role during Kras-induced tumorigenesis.We found that high expression of p130Cas is frequently detected in PDAC and correlates with higher histologic grade and poor prognosis. In a model of Kras-driven PDAC, loss of p130Cas inhibits tumor development and potently extends median survival. Deletion of p130Cas suppresses acinar-derived tumorigenesis and progression by means of repressing PI3K-AKT signaling, even in the presence of a worsening condition like pancreatitis.Our observations finally demonstrated that p130Cas acts downstream of Kras to boost the PI3K activity required for acinar to ductal metaplasia and subsequent tumor initiation. This demonstrates an unexpected driving role of p130Cas downstream of Kras through PI3K/AKT, thus indicating a rational therapeutic strategy of targeting the PI3K pathway in tumors with high expression of p130Cas.Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Product Citations: 20
In Gastroenterology on 1 April 2022 by Costamagna, A., Natalini, D., et al.
-
Cancer Research
ACK1 is dispensable for development, skin tumor formation, and breast cancer cell proliferation.
In FEBS Open Bio on 1 June 2021 by Brandao, R., Kwa, M. Q., et al.
Activated Cdc42-associated kinase 1 (ACK1), a widely expressed nonreceptor tyrosine kinase, is often amplified in cancer and has been shown to interact with Cell division cycle 42 (Cdc42), Epidermal growth factor receptor (EGFR), and several other cancer-relevant molecules, suggesting a possible role for ACK1 in development and tumor formation. To directly address this scenario, we generated mice lacking a functional ACK1 gene (ACK1 ko) using CRISPR genome editing. ACK1 ko mice developed normally, displayed no obvious defect in tissue maintenance, and were fertile. Primary ACK1-null keratinocytes showed normal phosphorylation of EGFR, but a tendency toward reduced activation of AKT serine/threonine kinase 1 (Akt) and Mitogen-activated protein kinase 1 (Erk). DMBA/TPA-induced skin tumor formation did not reveal significant differences between ACK1 ko and control mice. Deletion of the ACK1 gene in the breast cancer cell lines MDA-MB-231, 67NR, MCF7, 4T1, and T47D caused no differences in growth. Furthermore, EGF-induced phosphorylation kinetics of Erk, Akt, and p130Cas were not detectably altered in T47D cells by the loss of ACK1. Finally, loss of ACK1 in MDA-MB-231 and T47D breast cancer cells had a very limited or no effect on directed cell migration. These data do not support a major role for ACK1 in Cdc42 and EGFR signaling, development, or tumor formation.
© 2021 The Authors. FEBS Open Bio published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
-
WB
-
Cancer Research
In The Journal of Biological Chemistry on 10 February 2021 by Park, T., Large, N., et al.
The expression levels of CT10 regulator of kinase (Crk) and Crk-like (CrkL) are elevated in many human cancers, including glioblastoma (GBM), and are believed to contribute to poor prognosis. Although Crk and CrkL have been proposed as therapeutic targets in these tumors, the lack of a reliable, quantitative assay to measure Crk and CrkL activity has hindered development of inhibitors. Here, we knocked down Crk, CrkL, or both using siRNAs in a human GBM cell line, U-118MG, to determine the respective, quantitative contributions of Crk and CrkL to cellular phenotypes. The combined use of specific and potent Crk and CrkL siRNAs induced effective knockdown of CrkII, CrkI, and CrkL. Whereas Crk knockdown did not affect cell morphology, proliferation, adhesion, or invasion, CrkL knockdown caused shrinkage of cells and inhibition of cell proliferation, adhesion, and invasion. Crk/CrkL double knockdown resulted in more pronounced morphological alterations and more robust inhibition of proliferation, adhesion, and invasion. Furthermore, Crk/CrkL double knockdown completely blocked cell migration, and this effect was rescued by transient overexpression of CrkL but not of Crk. Quantification of protein levels indicated that CrkL is expressed more abundantly than CrkII and CrkI in U-118MG cells. These results demonstrate both the predominant role of CrkL and the essential overlapping functions of Crk and CrkL in U-118MG cells. Furthermore, our study indicates that migration of U-118MG cells depends entirely on Crk and CrkL. Thus, impedance-based, real-time measurement of tumor cell migration represents a robust assay for monitoring Crk and CrkL activities.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
Essential role of the Crk family-dosage in DiGeorge-like anomaly and metabolic homeostasis.
In Life Science Alliance on 1 February 2020 by Imamoto, A., Ki, S., et al.
CRK and CRKL (CRK-like) encode adapter proteins with similar biochemical properties. Here, we show that a 50% reduction of the family-combined dosage generates developmental defects, including aspects of DiGeorge/del22q11 syndrome in mice. Like the mouse homologs of two 22q11.21 genes CRKL and TBX1, Crk and Tbx1 also genetically interact, thus suggesting that pathways shared by the three genes participate in organogenesis affected in the syndrome. We also show that Crk and Crkl are required during mesoderm development, and Crk/Crkl deficiency results in small cell size and abnormal mesenchyme behavior in primary embryonic fibroblasts. Our systems-wide analyses reveal impaired glycolysis, associated with low Hif1a protein levels as well as reduced histone H3K27 acetylation in several key glycolysis genes. Furthermore, Crk/Crkl deficiency sensitizes MEFs to 2-deoxy-D-glucose, a competitive inhibitor of glycolysis, to induce cell blebbing. Activated Rapgef1, a Crk/Crkl-downstream effector, rescues several aspects of the cell phenotype, including proliferation, cell size, focal adhesions, and phosphorylation of p70 S6k1 and ribosomal protein S6. Our investigations demonstrate that Crk/Crkl-shared pathways orchestrate metabolic homeostasis and cell behavior through widespread epigenetic controls.
© 2020 Imamoto et al.
-
WB
-
Biochemistry and Molecular biology
-
Cell Biology
The SRCIN1/p140Cap adaptor protein negatively regulates the aggressiveness of neuroblastoma.
In Cell Death and Differentiation on 1 February 2020 by Grasso, S., Cangelosi, D., et al.
Neuroblastoma is the most common extra-cranial pediatric solid tumor, responsible for 13-15% of pediatric cancer death. Its intrinsic heterogeneity makes it difficult to target for successful therapy. The adaptor protein p140Cap/SRCIN1 negatively regulates tumor cell features and limits breast cancer progression. This study wish to assess if p140Cap is a key biological determinant of neuroblastoma outcome. RNAseq profiles of a large cohort of neuroblastoma patients show that SRCIN1 mRNA levels are an independent risk factor inversely correlated to disease aggressiveness. In high-risk patients, CGH+SNP microarray analysis of primary neuroblastoma identifies SRCIN1 as frequently altered by hemizygous deletion, copy-neutral loss of heterozygosity, or disruption. Functional experiments show that p140Cap negatively regulates Src and STAT3 signaling, affects anchorage-independent growth and migration, in vivo tumor growth and spontaneous lung metastasis formation. p140Cap also increases sensitivity of neuroblastoma cells to doxorubicin and etoposide treatment, as well as to a combined treatment with chemotherapy drugs and Src inhibitors. Our functional findings point to a causal role of p140Cap in curbing the aggressiveness of neuroblastoma, due to its ability to impinge on specific molecular pathways, and to sensitize cells to therapeutic treatment. This study provides the first evidence that the SRCIN1/p140Cap adaptor protein is a key player in neuroblastoma as a new independent prognostic marker for patient outcome and treatment. Altogether, these data highlight the potential clinical impact of SRCIN1/p140Cap expression in neuroblastoma tumors, in terms of reducing cytotoxic effects of chemotherapy, one of the main issues for pediatric tumor treatment.
-
WB
-
Cancer Research
-
Cell Biology
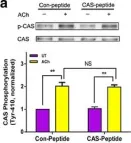
In J Biol Chem on 10 February 2021 by Park, T., Large, N., et al.
Fig.1.B

-
WB
-
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Biol Chem on 10 February 2021 by Park, T., Large, N., et al.
Fig.2.B

-
WB
-
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Biol Chem on 10 February 2021 by Park, T., Large, N., et al.
Fig.3.A

-
WB
-
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Biol Chem on 10 February 2021 by Park, T., Large, N., et al.
Fig.5.A

-
WB
-
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 9
In J Biol Chem on 10 February 2021 by Park, T., Large, N., et al.
Fig.7.A

-
WB
-
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 9
In Cell Death Differ on 1 February 2020 by Grasso, S., Cangelosi, D., et al.
Fig.3.B

-
WB
-
Collected and cropped from Cell Death Differ by CiteAb, provided under a CC-BY license
Image 1 of 9
In Respir Res on 5 January 2018 by Wang, Y., Rezey, A. C., et al.
Fig.3.D

-
WB
-
Collected and cropped from Respir Res by CiteAb, provided under a CC-BY license
Image 1 of 9
In Respir Res on 5 January 2018 by Wang, Y., Rezey, A. C., et al.
Fig.5.A

-
WB
-
Collected and cropped from Respir Res by CiteAb, provided under a CC-BY license
Image 1 of 9
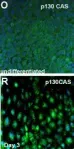
In BMC Genomics on 27 December 2007 by Schulz, T. C., Swistowska, A. M., et al.
Fig.4.O,R

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from BMC Genomics by CiteAb, provided under a CC-BY license
Image 1 of 9