p63 is a transcription factor with intrinsic pioneer factor activity and pleiotropic functions. Transforming growth factor β (TGFβ) signaling via activation and cooperative action of canonical, SMAD, and non-canonical, MAP-kinase (MAPK) pathways, elicits both anti- and pro-tumorigenic properties, including cell stemness and invasiveness. TGFβ activates the ΔNp63 transcriptional program in cancer cells; however, the link between TGFβ and p63 in unmasking the epigenetic landscape during tumor progression allowing chromatin accessibility and gene transcription, is not yet reported.
Small molecule inhibitors, including protein kinase inhibitors and RNA-silencing, provided loss of function analyses. Sphere formation assays in cancer cells, chromatin immunoprecipitation and mRNA expression assays were utilized in order to gain mechanistic evidence. Mass spectrometry analysis coupled to co-immunoprecipitation assays revealed novel p63 interactors and their involvement in p63-dependent transcription.
The sphere-forming capacity of breast cancer cells was enhanced upon TGFβ stimulation and significantly decreased upon ΔNp63 depletion. Activation of TGFβ signaling via p38 MAPK signaling induced ΔNp63 phosphorylation at Ser 66/68 resulting in stabilized ΔNp63 protein with enhanced DNA binding properties. TGFβ stimulation altered the ratio of H3K27ac and H3K27me3 histone modification marks, pointing towards higher H3K27ac and increased p300 acetyltransferase recruitment to chromatin. By silencing the expression of ΔNp63, the TGFβ effect on chromatin remodeling was abrogated. Inhibition of H3K27me3, revealed the important role of TGFβ as the upstream signal for guiding ΔNp63 to the TGFβ/SMAD gene loci, as well as the indispensable role of ΔNp63 in recruiting histone modifying enzymes, such as p300, to these genomic regions, regulating chromatin accessibility and gene transcription. Mechanistically, TGFβ through SMAD activation induced dissociation of ΔNp63 from NURD or NCOR/SMRT histone deacetylation complexes, while promoted the assembly of ΔNp63-p300 complexes, affecting the levels of histone acetylation and the outcome of ΔNp63-dependent transcription.
ΔNp63, phosphorylated and recruited by TGFβ to the TGFβ/SMAD/ΔNp63 gene loci, promotes chromatin accessibility and transcription of target genes related to stemness and cell invasion.
© 2024. The Author(s).
Product Citations: 25
In Cell Communication and Signaling : CCS on 23 August 2024 by Vasilaki, E., Bai, Y., et al.
-
Homo sapiens (Human)
-
Cancer Research
-
Endocrinology and Physiology
-
Genetics
Repair oligodendrocytes demyelinating and disintegrating damaged axons after injury
Preprint on BioRxiv : the Preprint Server for Biology on 18 May 2023 by Nocera, G., Vaquié, A., et al.
After a spinal cord injury, axons fail to regrow, which results in permanent loss of function 1 . This is in contrast with peripheral axons that can regrow efficiently after injury 2 . These differences are partly due to the different plasticity of myelinating cells, Schwann cells and oligodendrocytes, in these two systems 3 . The molecular mechanisms underlying this different plasticity remain however poorly understood. Here, we show that the phosphatase Dusp6 4 is a master inhibitor of oligodendrocyte plasticity after spinal cord injury. Dusp6 is rapidly downregulated in Schwann cells and upregulated in oligodendrocytes after axon injury. Simultaneously, the MAP kinases ERK1/2 are activated and the transcription factor c-Jun is upregulated in Schwann cells 5,6 , but not in oligodendrocytes. Ablation or inactivation of Dusp6 induces rapid ERK1/2 phosphorylation, c-Jun upregulation and filopodia formation in oligodendrocytes, leading to mechanically-induced, fast disintegration of distal ends of injured axons, myelin clearance and axonal regrowth. Together, our findings provide understanding of the mechanisms underlying the different plasticity of Schwann cells and oligodendrocytes after injury and a method to convert mature oligodendrocytes exhibiting inhibitory cues for axonal regrowth into repair oligodendrocytes reminiscent of repair Schwann cells. We show that repair oligodendrocytes successfully increase the compatibility of the spinal cord environment with axonal regrowth after injury, suggesting a potential use of repair oligodendrocytes as future therapeutic approach to treat spinal cord injuries.
-
Neuroscience
In NPJ Breast Cancer on 9 July 2021 by Chen, H., Padia, R., et al.
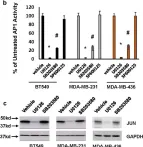
Triple negative breast cancer (TNBC) cells are generally more invasive than estrogen receptor-positive (ER + ) breast cancer cells. Consistent with the importance of activator protein 1 (AP1) transcription factors in invasion, AP1 activity is much higher in TNBC lines than ER + lines. In TNBC cells, robust AP1 activity is facilitated by both ERK and p38MAPK signaling pathways. While ERK signaling pathway regulates AP1 activity by controlling the abundance of AP1 transcription factors, p38MAPK signaling pathway does it by enhancing AP1 binding to AP1 sites without altering their abundance. Here, we show that p38MAPK regulation of AP1 activity involves both MAPKAPK2 (MK2) and JAB1, a known JUN-binding protein. MK2 not only interacts with JAB1 but also directly phosphorylates JAB1 at Ser177 in TNBC cells. Interestingly, Ser177 phosphorylation does not affect JAB1 and JUN interaction. Instead, interfering with p38MAPK signaling pathway or introducing an S to A point mutation at Ser177 of JAB1 reduces JUN recruitment to the AP1 sites in cyclin D1, urokinase plasminogen activator (uPA) and uPA receptor promoters. Moreover, knockdown of JAB1 diminishes >60% of AP1 transcriptional activity in TNBC cells. Taken together, these results indicate that MK2-mediated phosphorylation of JAB1 facilitates JUN recruitment to AP1 sites, thus augmenting AP1 activity. In line with the role of JAB1 in AP1 activity, silencing JAB1 leads to dramatic reduction in TNBC cell growth, in vitro invasion and in vivo tumor outgrowth. This study suggests that the p38MAPK-MK2 signaling pathway promotes TNBC tumorigenesis by sustaining robust AP1 activity.
© 2021. The Author(s).
-
ICC-IF
-
WB
-
Cancer Research
Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer.
In The EMBO Journal on 1 September 2020 by Feldker, N., Ferrazzi, F., et al.
Invasion, metastasis and therapy resistance are the major cause of cancer-associated deaths, and the EMT-inducing transcription factor ZEB1 is a crucial stimulator of these processes. While work on ZEB1 has mainly focused on its role as a transcriptional repressor, it can also act as a transcriptional activator. To further understand these two modes of action, we performed a genome-wide ZEB1 binding study in triple-negative breast cancer cells. We identified ZEB1 as a novel interactor of the AP-1 factors FOSL1 and JUN and show that, together with the Hippo pathway effector YAP, they form a transactivation complex, predominantly activating tumour-promoting genes, thereby synergising with its function as a repressor of epithelial genes. High expression of ZEB1, YAP, FOSL1 and JUN marks the aggressive claudin-low subtype of breast cancer, indicating the translational relevance of our findings. Thus, our results link critical tumour-promoting transcription factors: ZEB1, AP-1 and Hippo pathway factors. Disturbing their molecular interaction may provide a promising treatment option for aggressive cancer types.
© 2020 The Authors. Published under the terms of the CC BY NC ND 4.0 license.
-
IP
-
WB
-
EMSA
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cancer Research
In The Journal of Experimental Medicine on 2 March 2020 by Shibata, S., Kashiwagi, M., et al.
Keratinocytes respond to environmental signals by eliciting induction of genes that preserve skin's integrity. Here we show that the transcriptional response to stress signaling is supported by short-lived epigenetic changes. Comparison of chromatin accessibility and transcriptional changes induced by barrier disruption or by loss of the nucleosome remodeler Mi-2β identified their striking convergence in mouse and human keratinocytes. Mi-2β directly repressed genes induced by barrier disruption by restricting AP1-enriched promoter-distal sites, occupied by Mi-2β and JUNB at steady state and by c-JUN after Mi-2β depletion or stress signaling. Barrier disruption led to a modest reduction in Mi-2β expression and a further selective reduction of Mi-2β localization at stress response genes, possibly through competition with activated c-JUN. Consistent with a repressive role at stress response genes, genetic ablation of Mi-2β did not prevent reestablishment of barrier integrity but was required for return to homeostasis. Thus, a competition between Mi-2β-repressive and activating AP1 complexes may permit rapid transcriptional response to and resolution from stress signaling.
© 2019 Shibata et al.
-
IP
In NPJ Breast Cancer on 9 July 2021 by Chen, H., Padia, R., et al.
Fig.1.B

-
WB
-
Collected and cropped from NPJ Breast Cancer by CiteAb, provided under a CC-BY license
Image 1 of 7
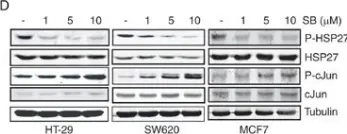
In Front Cell Neurosci on 12 November 2019 by Wu, L., Zeng, S., et al.
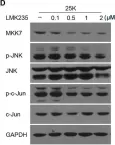
Fig.3.D

-
WB
-
Collected and cropped from Front Cell Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 7
In Front Cell Neurosci on 12 November 2019 by Wu, L., Zeng, S., et al.
Fig.3.E

-
WB
-
Collected and cropped from Front Cell Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 7
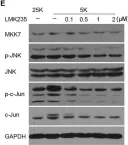
In Front Cell Neurosci on 12 November 2019 by Wu, L., Zeng, S., et al.
Fig.2.A

-
WB
-
Collected and cropped from Front Cell Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 7
In Front Cell Neurosci on 12 November 2019 by Wu, L., Zeng, S., et al.
Fig.2.D

-
WB
-
Collected and cropped from Front Cell Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 7
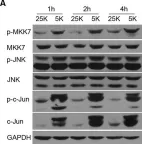
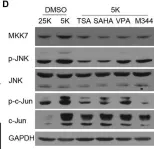
In EMBO Mol Med on 1 November 2013 by Pereira, L., Igea, A., et al.
Fig.1.D

-
WB
-
Collected and cropped from EMBO Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 7
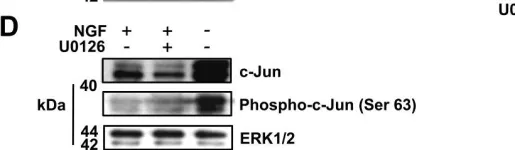
In BMC Neurosci on 15 July 2011 by Hughes, R., Gilley, J., et al.
Fig.1.D

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from BMC Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 7