

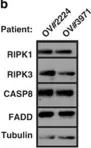
Autoimmune lymphoproliferative syndrome (ALPS) is a primary disorder of lymphocyte homeostasis, leading to chronic lymphoproliferation, autoimmune cytopenia, and increased risk of lymphoma. The genetic landscape of ALPS includes mutations in FAS, FASLG, and FADD, all associated with apoptosis deficiency, while the role of CASP10 defect in the disease remains debated. In this study, we aimed to assess the impact of CASP10 variants on ALPS pathogenesis. We benefit from thousands of genetic analysis datasets performed in our Institute's genetic platform to identify individuals carrying CASP10 variants previously suspected to be involved in ALPS outcome: p.C401LfsX15, p.V410I and p.Y446C, both at heterozygous and homozygous state. Clinical and laboratory features of the six included subjects were variable but not consistent with ALPS. Two individuals were healthy. Comprehensive analyses of CASP10 protein expression and FAS-mediated apoptosis were conducted and compared to healthy controls and ALPS patients with FAS mutations. Missense CASP10 variants (p.V410I and p.Y446C), which are common in the general population, did not disrupt CASP10 expression, nor FAS-mediated apoptosis. In contrast, homozygous p.C401LfsX15 CASP10 variant lead to a complete abolished CASP10 expression but had no impact on FAS-mediated apoptosis function. At heterozygous state, this p.C401LfsX15 variant lead to a reduced CASP10 protein levels but remained associated with a normal FAS-mediated apoptosis function. These findings demonstrate that CASPASE 10 is dispensable for FAS-mediated apoptosis. In consequences, CASP10 defect unlikely contribute to ALPS pathogenesis, since they did not result in an impairment of FAS-mediated apoptosis nor in clinical features of ALPS in human. Moreover, the absence of FAS expression up-regulation in subjects with CASP10 variants rule out any compensatory mechanisms possibly involved in the normal apoptosis function observed. In conclusion, this study challenges the notion that CASP10 variants contribute to the development of ALPS.
© 2024. The Author(s).
Product Citations: 41
In Cell Death & Disease on 4 May 2024 by Consonni, F., Moreno, S., et al.
-
Homo sapiens (Human)
-
Cell Biology
-
Immunology and Microbiology
In Experimental & Molecular Medicine on 1 April 2024 by Kim, J. H., Lee, J., et al.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a promising anticancer agent because it kills cancer cells while sparing normal cells. However, many cancers, including pancreatic ductal adenocarcinoma (PDAC), exhibit intrinsic or acquired resistance to TRAIL, and the molecular mechanisms underlying TRAIL resistance in cancers, particularly in PDAC, remain unclear. In this study, we demonstrated that glutamine (Gln) endows PDAC cells with resistance to TRAIL through KDM4C-mediated epigenetic regulation of cFLIP. Inhibition of glutaminolysis significantly reduced the cFLIP level, leading to TRAIL-mediated formation of death-inducing signaling complexes. Overexpression of cFLIP dramatically rescued PDAC cells from TRAIL/Gln deprivation-induced apoptosis. Alpha-Ketoglutarate (aKG) supplementation significantly reversed the decrease in the cFLIP level induced by glutaminolysis inhibition and rescued PDAC cells from TRAIL/Gln deprivation-induced apoptosis. Knockdown of glutamic-oxaloacetic transaminase 2, which facilitates the conversion of oxaloacetate and glutamate into aspartate and aKG, decreased aKG production and the cFLIP level and activated TRAIL-induced apoptosis. AKG-mediated epigenetic regulation was necessary for maintaining a high level of cFLIP. Glutaminolysis inhibition increased the abundance of H3K9me3 in the cFLIP promoter, indicating that Gln-derived aKG production is important for Jumonji-domain histone demethylase (JHDM)-mediated cFLIP regulation. The JHDM KDM4C regulated cFLIP expression by binding to its promoter, and KDM4C knockdown sensitized PDAC cells to TRAIL-induced apoptosis. The present findings suggest that Gln-derived aKG production is required for KDM4C-mediated epigenetic regulation of cFLIP, which leads to resistance to TRAIL.
© 2024. The Author(s).
-
Biochemistry and Molecular biology
-
Cancer Research
-
Genetics
Preprint on BioRxiv : the Preprint Server for Biology on 27 December 2021 by Oikawa, D., Gi, M., et al.
Deubiquitylating enzymes (DUBs) regulate numerous cellular functions by removing ubiquitin modifications. We examined the effects of 88 human DUBs on linear ubiquitin chain assembly complex (LUBAC)-induced NF-κB activation, and identified OTUD1 as a potent suppressor. OTUD1 regulates the canonical NF-κB pathway by hydrolysing K63-linked ubiquitin chains from NF-κB signalling factors, including LUBAC. OTUD1 negatively regulates the canonical NF-κB activation, apoptosis, and necroptosis, whereas OTUD1 upregulates the interferon (IFN) antiviral pathway. The N-terminal intrinsically disordered region of OTUD1, which contains an EGTE motif, is indispensable for KEAP1-binding and NF-κB suppression. OTUD1 is involved in the KEAP1-mediated antioxidant response and reactive oxygen species (ROS)-induced cell death, oxeiptosis. In Otud1 -/- -mice, inflammation, oxidative damage, and cell death were enhanced in inflammatory bowel disease, acute hepatitis, and sepsis models. Thus, OTUD1 is a crucial regulator for the inflammatory, innate immune, and oxidative stress responses and ROS-associated cell death pathways.
-
Immunology and Microbiology
Noncompetitive Allosteric Antagonism of Death Receptor 5 by a Synthetic Affibody Ligand.
In Biochemistry on 13 October 2020 by Vunnam, N., Szymonski, S., et al.
Fatty acid-induced upregulation of death receptor 5 (DR5) and its cognate ligand, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), promotes hepatocyte lipoapoptosis, which is a key mechanism in the progression of fatty liver disease. Accordingly, inhibition of DR5 signaling represents an attractive strategy for treating fatty liver disease. Ligand competition strategies are prevalent in tumor necrosis factor receptor antagonism, but recent studies have suggested that noncompetitive inhibition through perturbation of the receptor conformation may be a compelling alternative. To this end, we used yeast display and a designed combinatorial library to identify a synthetic 58-amino acid affibody ligand that specifically binds DR5. Biophysical and biochemical studies show that the affibody neither blocks TRAIL binding nor prevents the receptor-receptor interaction. Live-cell fluorescence lifetime measurements indicate that the affibody induces a conformational change in transmembrane dimers of DR5 and favors an inactive state of the receptor. The affibody inhibits apoptosis in TRAIL-treated Huh-7 cells, an in vitro model of fatty liver disease. Thus, this lead affibody serves as a potential drug candidate, with a unique mechanism of action, for fatty liver disease.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
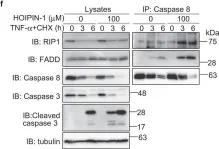
Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses.
In Communications Biology on 3 April 2020 by Oikawa, D., Sato, Y., et al.
The NF-κB and interferon antiviral signaling pathways play pivotal roles in inflammatory and innate immune responses. The LUBAC ubiquitin ligase complex, composed of the HOIP, HOIL-1L, and SHARPIN subunits, activates the canonical NF-κB pathway through Met1-linked linear ubiquitination. We identified small-molecule chemical inhibitors of LUBAC, HOIPIN-1 and HOIPIN-8. Here we show that HOIPINs down-regulate not only the proinflammatory cytokine-induced canonical NF-κB pathway, but also various pathogen-associated molecular pattern-induced antiviral pathways. Structural analyses indicated that HOIPINs inhibit the RING-HECT-hybrid reaction in HOIP by modifying the active Cys885, and residues in the C-terminal LDD domain, such as Arg935 and Asp936, facilitate the binding of HOIPINs to LUBAC. HOIPINs effectively induce cell death in activated B cell-like diffuse large B cell lymphoma cells, and alleviate imiquimod-induced psoriasis in model mice. These results reveal the molecular and cellular bases of LUBAC inhibition by HOIPINs, and demonstrate their potential therapeutic uses.
-
WB
-
Homo sapiens (Human)
-
Immunology and Microbiology
In Commun Biol on 3 April 2020 by Oikawa, D., Sato, Y., et al.
Fig.5.F

-
WB
-
Homo sapiens (Human)
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In Front Cell Dev Biol on 11 February 2020 by Wang, L., Chang, X., et al.
Fig.2.A

-
WB
-
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In Nat Commun on 24 August 2016 by Nakazawa, S., Oikawa, D., et al.
Fig.5.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
In Nat Commun on 24 August 2016 by Nakazawa, S., Oikawa, D., et al.
Fig.5.E

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
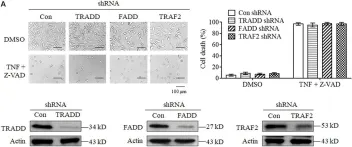
In Cell Death Dis on 30 October 2014 by McCabe, K. E., Bacos, K., et al.
Fig.3.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 7
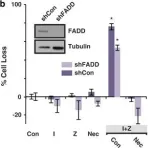
In Cell Death Dis on 30 October 2014 by McCabe, K. E., Bacos, K., et al.
Fig.1.C

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 7
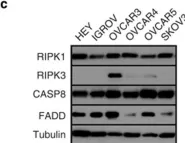
In Cell Death Dis on 30 October 2014 by McCabe, K. E., Bacos, K., et al.
Fig.6.B

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 7