
Biomolecular condensates are thought to create subcellular microenvironments that have different physicochemical properties compared with their surrounding nucleoplasm or cytoplasm1-5. However, probing the microenvironments of condensates and their relationship to biological function is a major challenge because tools to selectively manipulate specific condensates in living cells are limited6-9. Here, we develop a non-natural micropeptide (that is, the killswitch) and a nanobody-based recruitment system as a universal approach to probe endogenous condensates, and demonstrate direct links between condensate microenvironments and function in cells. The killswitch is a hydrophobic, aromatic-rich sequence with the ability to self-associate, and has no homology to human proteins. When recruited to endogenous and disease-specific condensates in human cells, the killswitch immobilized condensate-forming proteins, leading to both predicted and unexpected effects. Targeting the killswitch to the nucleolar protein NPM1 altered nucleolar composition and reduced the mobility of a ribosomal protein in nucleoli. Targeting the killswitch to fusion oncoprotein condensates altered condensate compositions and inhibited the proliferation of condensate-driven leukaemia cells. In adenoviral nuclear condensates, the killswitch inhibited partitioning of capsid proteins into condensates and suppressed viral particle assembly. The results suggest that the microenvironment within cellular condensates has an essential contribution to non-stoichiometric enrichment and mobility of effector proteins. The killswitch is a widely applicable tool to alter the material properties of endogenous condensates and, as a consequence, to probe functions of condensates linked to diverse physiological and pathological processes.
© 2025. The Author(s).
Product Citations: 222
Probing condensate microenvironments with a micropeptide killswitch.
In Nature on 4 June 2025 by Zhang, Y., Stöppelkamp, I., et al.
In Nature Communications on 26 May 2025 by Kawamura, A., Fujii, K., et al.
Mutations in the gene encoding chromodomain helicase DNA-binding protein 8 (CHD8) are strongly associated with autism spectrum disorder (ASD). Although duplications of the chromosomal locus including CHD8 have also been detected in individuals with neurodevelopmental disorders, the contribution of CHD8 duplication to clinical phenotypes and the underlying mechanisms have remained unknown. Here we show that Chd8 knock-in (KI) mice that overexpress CHD8 as a model of human CHD8 duplication manifest growth retardation, microcephaly, impaired neuronal differentiation, and behavioral abnormalities including hyperactivity and reduced anxiety-like behavior. Chd8 overexpression affects the transcription and chromatin accessibility of genes related to neurogenesis, with these changes being associated with aberrant binding of CHD8 to enhancer regions. Furthermore, pharmacological intervention partially ameliorates the hyperactivity of Chd8 KI mice. Our results thus indicate that Chd8 KI mice recapitulate key features of CHD8 duplication syndrome in humans, providing insight into pathogenic mechanisms underlying neurodevelopmental disorders.
© 2025. The Author(s).
-
WB
-
Mus musculus (House mouse)
-
Neuroscience
In Molecular Imaging on 24 April 2025 by Bini, J., Strober, J., et al.
In rodents, 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) catalyzes the conversion of inactive 11-dehydrocorticosterone to the active hormone corticosterone. Dysregulation of intracellular glucocorticoid action is implicated in metabolic diseases. Assessing 11β-HSD1 enzyme levels in vivo may be key to understanding obesity pathophysiology.
We used a Zucker Fatty (ZF) rat model and [18F]AS2471907 PET imaging to determine appropriate kinetic modeling methods and assess changes in 11β-HSD1 levels due to obesity in the liver, white and brown adipose tissue (WAT/BAT), and brain.
To validate [18F]AS2471907 PET in preclinical models, time-activity curves (TACs) were generated and kinetic modeling was performed with image-derived input functions (IDIFs) extracted from multiple locations. Quantitative estimates of radioligand binding were compared with ex vivo 11β-HSD1 protein expression. Validated quantitative PET kinetic modeling methods were then used to assess differences in 11β-HSD1 between lean and obese ZF rats. Metabolic disease status was confirmed with stable isotopes tracer studies of glucose and fatty acid metabolism.
Obesity is associated with decreased brain 11β-HSD1 levels, measured by [18F]AS2471907 PET, which correlated with measures of glucose and fatty acid metabolism.
We demonstrate that [18F]AS2471907 PET can provide useful quantification of 11β-HSD1 levels in a rodent model of obesity.
© The Author(s) 2024.
Preprint on BioRxiv : the Preprint Server for Biology on 6 February 2025 by Ma, R., Gong, L., et al.
ABSTRACT Background The liver undergoes significant hemodynamic changes during surgery, transplantation, or cirrhosis with portal hypertension(PH). The hepatic artery buffer response(HABR), which compensates for reduced portal venous flow by increasing hepatic artery(HA) flow, is hypothesized to induce pathological portal tract remodeling. This study investigates the molecular mechanisms underlying this process. Methods PH was induced in Sprague-Dawley rats via partial portal vein ligation(PPVL). Structural evaluation(microCT), immune cell profiling, hemodynamic measurements, and transcriptomic analysis in macrophages(Mϕ) from sham or PPVL rats were conducted. Results MicroCT revealed decreased portal vein flow and increased HA flow correlated with portal pressure(r=0.799, p<0.01). A 2-fold increase in portal tract fibrosis(p<0.001) was observed with increased α-SMA+ myofibroblasts in PPVL rats. CD68+ Mϕ peaked at 10 days post-PPVL, and their depletion significantly reduced fibrosis(p<0.001), indicating critical roles of Mϕ in portal tract remodeling. VCAM-1 was elevated in HA endothelium and portal fibroblasts (PFs); VCAM-1 neutralization reduced collagen accumulation(p<0.05), CD68+ Mϕ(46.3%, p<0.01), and CD3+ T cells(18%, p<0.05). Mϕ-conditioned medium increased VCAM-1 in PFs(8-fold, p<0.001) and enhanced PF migration, while VCAM-1 knockdown reduced this effect (p<0.01). Single-cell RNA sequencing data(GSE171904) and RNA-FISH revealed increased interactions between osteopontin (Spp1)+ Mϕ and PFs, with Spp1+ Mϕ driving fibrosis. Spp1 knockdown in Mϕ co-culture reduced PF fibrogenic markers, while recombinant Spp1 upregulated Col1a1, Fn1, and Acta2 expression in PFs. Conclusion Increased VCAM-1 in arterial endothelial cells and PFs facilitates the recruitment of Spp1+ Mϕ, which drive HA flow-mediated vascular remodeling and portal tract fibrosis. These findings highlight arterial flow-induced fibrosis as a key mechanism in PH, potentially contributing to disease progression and decompensation. Synopsis Liver hemodynamic changes in portal hypertension drive extracellular matrix accumulation and portal tract remodeling via Spp1+ macrophages. This study highlights how altered blood flow induces fibrosis, and its potential role in decompensation, and identifies therapeutic targets for advanced liver disease.
-
Cardiovascular biology
In Nature Communications on 1 February 2025 by Van den Bossche, V., Vignau, J., et al.
Anti-epidermal growth factor receptor (EGFR) therapy (cetuximab) shows a limited clinical benefit for patients with locally advanced or recurrent/metastatic head and neck squamous cell carcinoma (HNSCC), due to the frequent occurrence of secondary resistance mechanisms. Here we report that cetuximab-resistant HNSCC cells display a peroxisome proliferator-activated receptor alpha (PPARα)-mediated lipid metabolism reprogramming, with increased fatty acid uptake and oxidation capacities, while glycolysis is not modified. This metabolic shift makes cetuximab-resistant HNSCC cells particularly sensitive to a pharmacological inhibition of either carnitine palmitoyltransferase 1A (CPT1A) or PPARα in 3D spheroids and tumor xenografts in mice. Importantly, the PPARα-related gene signature, in human clinical datasets, correlates with lower response to anti-EGFR therapy and poor survival in HNSCC patients, thereby validating its clinical relevance. This study points out lipid metabolism rewiring as a non-genetic resistance-causing mechanism in HNSCC that may be therapeutically targeted to overcome acquired resistance to anti-EGFR therapy.
© 2025. The Author(s).
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cancer Research
-
Cell Biology
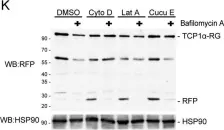
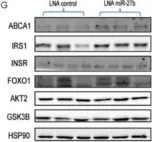
In Nat Commun on 16 March 2024 by Zhao, Y., Ning, J., et al.
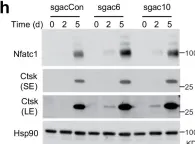
Fig.7.H

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 65
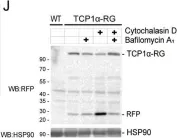
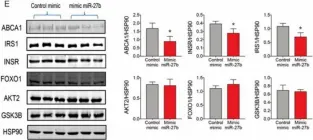
In Nat Commun on 16 March 2024 by Zhao, Y., Ning, J., et al.
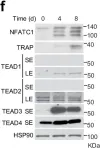
Fig.6.F

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 65
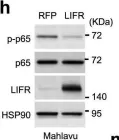
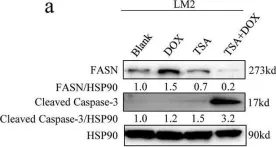
In Nat Commun on 16 March 2024 by Zhao, Y., Ning, J., et al.
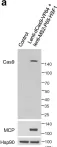
Fig.7.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 65
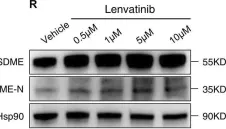
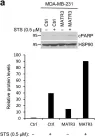
In Cell Death Dis on 17 July 2023 by He, Y., Zhang, Q., et al.
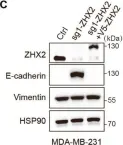
Fig.2.C

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
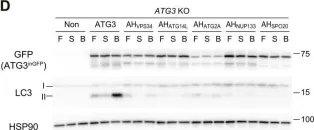
In Sci Adv on 23 June 2023 by Nishimura, T., Lazzeri, G., et al.
Fig.1.D

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 65
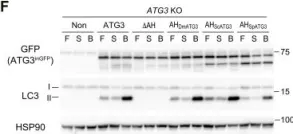
In Sci Adv on 23 June 2023 by Nishimura, T., Lazzeri, G., et al.
Fig.1.F

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 65
In Sci Adv on 23 June 2023 by Nishimura, T., Lazzeri, G., et al.
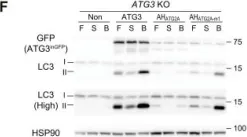
Fig.3.F

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 65
In Front Cardiovasc Med on 22 November 2022 by Aranda, J. F., Pérez-García, A., et al.
Fig.6.B

-
WB
-
Collected and cropped from Front Cardiovasc Med by CiteAb, provided under a CC-BY license
Image 1 of 65
In Front Cardiovasc Med on 22 November 2022 by Aranda, J. F., Pérez-García, A., et al.
Fig.7.B

-
WB
-
Collected and cropped from Front Cardiovasc Med by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cells on 16 August 2022 by Martín-Martín, Y., Pérez-García, A., et al.
Fig.5.B

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 65
In Elife on 8 August 2022 by Yim, W. W., Yamamoto, H., et al.
Fig.1.A

-
WB
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 65
In Elife on 8 August 2022 by Yim, W. W., Yamamoto, H., et al.
Fig.4.A

-
WB
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Discov on 25 January 2022 by Date, Y., Matsuura, A., et al.
Fig.2.K

-
WB
-
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Discov on 25 January 2022 by Date, Y., Matsuura, A., et al.
Fig.2.J

-
WB
-
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 65
In Nat Commun on 17 December 2021 by Yao, F., Deng, Y., et al.
Fig.3.H

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 65
In Front Cell Dev Biol on 26 November 2021 by Fu, X. W. & Song, C. Q.
Fig.11.R

-
WB
-
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 30 August 2021 by Luo, H., Zhou, Z., et al.
Fig.7.A

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
In Int J Mol Sci on 17 November 2020 by Benito-Vicente, A., Uribe, K. B., et al.
Fig.2.E

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 65
In Int J Mol Sci on 17 November 2020 by Benito-Vicente, A., Uribe, K. B., et al.
Fig.4.G

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 65
In Biol Res on 25 September 2020 by Yang, J., Lee, S. J., et al.
Fig.3.A

-
WB
-
Collected and cropped from Biol Res by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 4 May 2020 by Wu, Y. H., Chou, T. F., et al.
Fig.5.C

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 4 May 2020 by Wu, Y. H., Chou, T. F., et al.
Fig.5.D

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 4 May 2020 by Wu, Y. H., Chou, T. F., et al.
Fig.5.F

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 4 May 2020 by Wu, Y. H., Chou, T. F., et al.
Fig.6.E

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65
In Cell Death Dis on 4 May 2020 by Wu, Y. H., Chou, T. F., et al.
Fig.6.C

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 65