

The neuromuscular junction (NMJ) is the unique interface between lower motor neurons and skeletal muscle fibers and is indispensable for muscle function. Tight control of its localized formation at the center of every muscle fiber, and maintenance throughout lifetime are sustained by muscle-specific kinase (MuSK). MuSK acts as central regulator of acetylcholine receptor clustering at the postsynapse. Localized and temporally controlled signaling of MuSK is primarily achieved by tyrosine autophosphorylation and inhibition thereof. Previous investigations suggested serine phosphorylation of the activation domain as an additional modulator of MuSK activation. Here we identified calcium/calmodulin dependent protein kinase II (CaMK2) and in particular CaMK2β as novel catalyst of MuSK activation and confirmed its capability to phosphorylate MuSK in heterologous cells. However, whereas CaMK2β absence in muscle cells reduced AChR clustering, MuSK phosphorylation was unchanged. Accordingly, we ruled out MuSK phosphorylation as the cause of synapse fragmentation in a mouse model for myotonic dystrophy type 1, in which the muscle-specific splice-variant of CaMK2β is missing, or as the cause of ataxia or delayed muscle development in CaMK2β knockout animals. Histological characterization of muscles of CaMK2β knockout mice indicated specific roles of CaMK2β in fast glycolytic versus slow oxidative muscle. Taken together, our data shows that MuSK can be phosphorylated by CaMK2β, but loss of CaMK2β is likely compensated by other CaMK2 paralogs at the NMJ.
© 2025. The Author(s).
Product Citations: 99
MuSK is a substrate for CaMK2β but this interaction is dispensable for MuSK activation in vivo.
In Scientific Reports on 28 April 2025 by Prömer, J. J., Wolske, S., et al.
MuSK is a substrate for CaMK2β but this interaction is dispensable for MuSK activation in vivo
Preprint on Research Square on 13 December 2024 by Prömer, J. J., Wolske, S., et al.
Abstract The neuromuscular junction (NMJ) is the unique interface between lower motor neurons and skeletal muscle fibers and is indispensable for muscle function. Tight control of its localized formation at the center of every muscle fiber, and maintenance throughout lifetime are sustained by muscle-specific kinase (MuSK). MuSK acts as central regulator of acetylcholine receptor clustering at the postsynapse. Localized and temporally controlled signaling of MuSK is primarily achieved by tyrosine autophosphorylation and inhibition thereof. Previous research suggested serine phosphorylation of the activation domain as additional modulator of MuSK activation. Here we identified calcium/calmodulin dependent protein kinase II (CaMK2) and in particular CaMK2β as novel catalyst of MuSK activation and confirmed its capability to phosphorylate MuSK in heterologous cells. However, whereas CaMK2β absence in muscle cells reduced AChR clustering, MuSK phosphorylation was unchanged. Accordingly, we ruled out MuSK phosphorylation as the cause of synapse fragmentation in a mouse model for myotonic dystrophy type 1, in which the muscle-specific splice-variant of CaMK2β is missing, or as the cause of ataxia or delayed muscle development in CaMK2β knockout animals. Histological characterization of muscles of CaMK2β knockout mice indicated specific roles of CaMK2β in fast glycolytic versus slow oxidative muscle. Taken together our data shows that MuSK can be phosphorylated by CaMK2b, but loss of CaMK2b is likely compensated for by other CaMK2 paralogs at the NMJ.
Unveiling the native architecture of adult cardiac tissue using the 3D-NaissI method
Preprint on BioRxiv : the Preprint Server for Biology on 28 November 2024 by Pataluch, N., Guilbeau-Frugier, C., et al.
Abstract Accurately imaging adult cardiac tissue in its native state is essential for regenerative medicine and understanding heart disease. Current fluorescence methods encounter challenges with tissue fixation. Here, we introduce the 3D-NaissI (3D-Native Tissue Imaging) method, enabling rapid, cost-effective imaging of fresh cardiac tissue samples in their closest native state, that we also extended to other tissues. We validated 3D-NaissI’s efficacy in preserving cardiac tissue integrity using small biopsies under hypothermic conditions in phosphate-buffered saline, offering unparalleled resolution in confocal microscopy for imaging fluorescent-small molecules/-antibodies. Compared to conventional histology, 3D-NaissI preserves cardiac tissue architecture and native protein epitopes, facilitating the use of a wide range of commercial antibodies without unmasking strategies. We successfully identified specific cardiac protein expression patterns in cardiomyocytes (CMs) from rodents and humans, including for the first time ACE2 localization in the lateral membrane/T-Tubules and SGTL2 in the sarcoplasmic reticulum. These findings shed light on COVID-19-related cardiac complications and suggest novel explanations for iSGLT2 therapeutic benefits in HFpEF patients. Additionally, we challenge the notion of "connexin-43 lateralization” in heart pathology, suggesting it may be an artifact of cardiac fixation, as 3D-NaissI clearly revealed native connexin-43 expression at the lateral membrane of healthy CMs. We also discovered previously undocumented periodic ring-like 3D structures formed by pericytes covering CMs’ lateral surfaces. These structures, positive for laminin-2, delineate a specific spatial architecture of laminin-2 receptors at the CM surface, highlighting the pivotal role of pericytes in CM function. Lastly, 3D-NaissI facilitates mapping native human protein expression in fresh cardiac autopsies, providing insights into both pathological and non-pathological contexts. Hence, 3D-NaissI offers unparalleled insights into native cardiac tissue biology and promises to advance our understanding of physiology and pathophysiology, surpassing standard histology in resolution and accuracy.
-
Cardiovascular biology
Palmitoylation regulates the magnitude of HCN4-mediated currents in mammalian cells.
In Frontiers in Physiology on 1 May 2023 by Congreve, S. D., Main, A., et al.
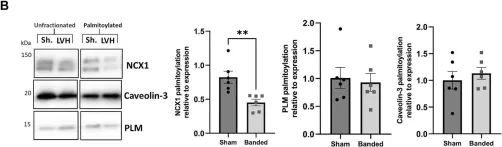
The sinoatrial node (SAN) and subsidiary pacemakers in the cardiac conduction system generate spontaneous electrical activity which is indispensable for electrical and therefore contractile function of the heart. The hyperpolarisation-activated cyclic nucleotide-gated channel HCN4 is responsible for genesis of the pacemaker "funny" current during diastolic depolarisation. S-palmitoylation, the reversible conjugation of the fatty acid palmitate to protein cysteine sulfhydryls, regulates the activity of key cardiac Na+ and Ca2+ handling proteins, influencing their membrane microdomain localisation and function. We investigated HCN4 palmitoylation and its functional consequences in engineered human embryonic kidney 293T cells as well as endogenous HCN4 in neonatal rat ventricular myocytes. HCN4 was palmitoylated in all experimental systems investigated. We mapped the HCN4 palmitoylation sites to a pair of cysteines in the HCN4 intracellular amino terminus. A double cysteine-to-alanine mutation CC93A/179AA of full length HCN4 caused a ∼67% reduction in palmitoylation in comparison to wild type HCN4. We used whole-cell patch clamp to evaluate HCN4 current (IHCN4) in stably transfected 293T cells. Removal of the two N-terminal palmitoylation sites did not significantly alter half maximal activation voltage of IHCN4 or the activation slope factor. IHCN4 was significantly larger in cells expressing wild type compared to non-palmitoylated HCN4 across a range of voltages. Phylogenetic analysis revealed that although cysteine 93 is widely conserved across all classes of HCN4 vertebrate orthologs, conservation of cysteine 179 is restricted to placental mammals. Collectively, we provide evidence for functional regulation of HCN4 via palmitoylation of its amino terminus in vertebrates. We suggest that by recruiting the amino terminus to the bilayer, palmitoylation enhances the magnitude of HCN4-mediated currents, but does not significantly affect the kinetics.
Copyright © 2023 Congreve, Main, Butler, Gao, Brown, Du, Choisy, Cheng, Hancox and Fuller.
-
WB
-
Endocrinology and Physiology
In eLife on 17 January 2023 by Karsenty, C., Guilbeau-Frugier, C., et al.
The rod-shaped adult cardiomyocyte (CM) harbors a unique architecture of its lateral surface with periodic crests, relying on the presence of subsarcolemmal mitochondria (SSM) with unknown role. Here, we investigated the development and functional role of CM crests during the postnatal period. We found in rodents that CM crest maturation occurs late between postnatal day 20 (P20) and P60 through both SSM biogenesis, swelling and crest-crest lateral interactions between adjacent CM, promoting tissue compaction. At the functional level, we showed that the P20-P60 period is dedicated to the improvement of relaxation. Interestingly, crest maturation specifically contributes to an atypical CM hypertrophy of its short axis, without myofibril addition, but relying on CM lateral stretching. Mechanistically, using constitutive and conditional CM-specific knock-out mice, we identified ephrin-B1, a lateral membrane stabilizer, as a molecular determinant of P20-P60 crest maturation, governing both the CM lateral stretch and the diastolic function, thus highly suggesting a link between crest maturity and diastole. Remarkably, while young adult CM-specific Efnb1 KO mice essentially exhibit an impairment of the ventricular diastole with preserved ejection fraction and exercise intolerance, they progressively switch toward systolic heart failure with 100% KO mice dying after 13 months, indicative of a critical role of CM-ephrin-B1 in the adult heart function. This study highlights the molecular determinants and the biological implication of a new late P20-P60 postnatal developmental stage of the heart in rodents during which, in part, ephrin-B1 specifically regulates the maturation of the CM surface crests and of the diastolic function.
© 2023, Karsenty et al.
In Front Physiol on 25 October 2022 by Main, A., Boguslavskyi, A., et al.
Fig.2.B

-
WB
-
Collected and cropped from Front Physiol by CiteAb, provided under a CC-BY license
Image 1 of 12
In Front Endocrinol (Lausanne) on 28 May 2019 by Kretschmar, C., Peña-Oyarzún, D., et al.
Fig.3.C

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 12
In Front Endocrinol (Lausanne) on 28 May 2019 by Kretschmar, C., Peña-Oyarzún, D., et al.
Fig.2.C

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 12
In Front Endocrinol (Lausanne) on 28 May 2019 by Kretschmar, C., Peña-Oyarzún, D., et al.
Fig.1.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 12
In Front Endocrinol (Lausanne) on 28 May 2019 by Kretschmar, C., Peña-Oyarzún, D., et al.
Fig.2.F

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Front Endocrinol (Lausanne) by CiteAb, provided under a CC-BY license
Image 1 of 12
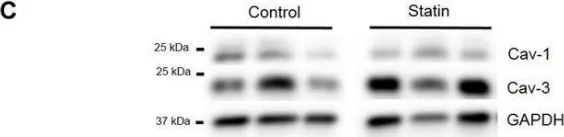
In Exp Physiol on 1 May 2019 by Kong, C. H. T., Bryant, S. M., et al.
Fig.3.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Exp Physiol by CiteAb, provided under a CC-BY license
Image 1 of 12
In Am J Physiol Heart Circ Physiol on 1 November 2018 by Bryant, S. M., Kong, C. H. T., et al.
Fig.1.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Am J Physiol Heart Circ Physiol by CiteAb, provided under a CC-BY license
Image 1 of 12
In Front Pharmacol on 5 May 2017 by Macdougall, D. A., Pugh, S. D., et al.
Fig.9.C

-
WB
-
Collected and cropped from Front Pharmacol by CiteAb, provided under a CC-BY license
Image 1 of 12
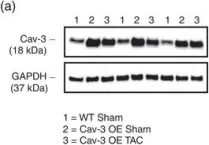
In PLoS One on 19 June 2015 by Faggi, F., Codenotti, S., et al.
Fig.4.B

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 12
In PLoS One on 19 June 2015 by Faggi, F., Codenotti, S., et al.
Fig.4.C

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 12
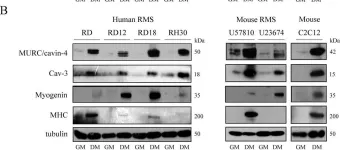
In PLoS One on 19 June 2015 by Faggi, F., Codenotti, S., et al.
Fig.5.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 12
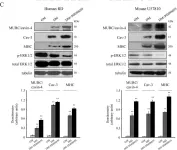
In PLoS One on 19 June 2015 by Faggi, F., Codenotti, S., et al.
Fig.6.D

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 12