

NDC80 complex (NDC80C) is composed of four subunits (SPC24, SPC25, NDC80, and NUF2) and is vital for kinetochore-microtubule (KT-MT) attachment during mitosis. Paradoxically, NDC80C also functions in the activation of the spindle-assembly checkpoint (SAC). This raises an interesting question regarding how mitosis is regulated when NDC80C levels are compromised. Using a degron-mediated depletion system, we found that acute silencing of SPC24 triggered a transient mitotic arrest followed by mitotic slippage. SPC24-deficient cells were unable to sustain SAC activation despite the loss of KT-MT interaction. Intriguingly, our results revealed that other subunits of the NDC80C were co-downregulated with SPC24 at a posttranslational level. Silencing any individual subunit of NDC80C likewise reduced the expression of the entire complex. We found that the SPC24-SPC25 and NDC80-NUF2 subcomplexes could be individually stabilized using ectopically expressed subunits. The synergism of SPC24 downregulation with drugs that promote either mitotic arrest or mitotic slippage further underscored the dual roles of NDC80C in KT-MT interaction and SAC maintenance. The tight coordinated regulation of NDC80C subunits suggests that targeting individual subunits could disrupt mitotic progression and provide new avenues for therapeutic intervention.
These results highlight the tight coordinated regulation of NDC80C subunits and their potential as targets for antimitotic therapies.
©2024 The Authors; Published by the American Association for Cancer Research.
Product Citations: 51
In Molecular Cancer Research on 2 May 2024 by Kim, S. H., Lau, T. Y., et al.
-
Cancer Research
-
Cell Biology
Alternative CDC20 translational isoforms tune mitotic arrest duration.
In Nature on 1 May 2023 by Tsang, M. J. & Cheeseman, I. M.
Mitotic defects activate the spindle-assembly checkpoint, which inhibits the anaphase-promoting complex co-activator CDC20 to induce a prolonged cell cycle arrest1,2. Once errors are corrected, the spindle-assembly checkpoint is silenced, allowing anaphase onset to occur. However, in the presence of persistent unresolvable errors, cells can undergo 'mitotic slippage', exiting mitosis into a tetraploid G1 state and escaping the cell death that results from a prolonged arrest. The molecular logic that enables cells to balance these duelling mitotic arrest and slippage behaviours remains unclear. Here we demonstrate that human cells modulate the duration of their mitotic arrest through the presence of conserved, alternative CDC20 translational isoforms. Downstream translation initiation results in a truncated CDC20 isoform that is resistant to spindle-assembly-checkpoint-mediated inhibition and promotes mitotic exit even in the presence of mitotic perturbations. Our study supports a model in which the relative levels of CDC20 translational isoforms control the duration of mitotic arrest. During a prolonged mitotic arrest, new protein synthesis and differential CDC20 isoform turnover create a timer, with mitotic exit occurring once the truncated Met43 isoform achieves sufficient levels. Targeted molecular changes or naturally occurring cancer mutations that alter CDC20 isoform ratios or its translational control modulate mitotic arrest duration and anti-mitotic drug sensitivity, with potential implications for the diagnosis and treatment of human cancers.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
-
Biochemistry and Molecular biology
-
Cell Biology
In The Journal of Biological Chemistry on 1 March 2023 by Ng, L. Y., Ma, H. T., et al.
Cyclin A and CDC25A are both activators of cyclin-dependent kinases (CDKs): cyclin A acts as an activating subunit of CDKs and CDC25A a phosphatase of the inhibitory phosphorylation sites of the CDKs. In this study, we uncovered an inverse relationship between the two CDK activators. As cyclin A is an essential gene, we generated a conditional silencing cell line using a combination of CRISPR-Cas9 and degron-tagged cyclin A. Destruction of cyclin A promoted an acute accumulation of CDC25A. The increase of CDC25A after cyclin A depletion occurred throughout the cell cycle and was independent on cell cycle delay caused by cyclin A deficiency. Moreover, we determined that the inverse relationship with cyclin A was specific for CDC25A and not for other CDC25 family members or kinases that regulate the same sites in CDKs. Unexpectedly, the upregulation of CDC25A was mainly caused by an increase in transcriptional activity instead of a change in the stability of the protein. Reversing the accumulation of CDC25A severely delayed G2-M in cyclin A-depleted cells. Taken together, these data provide evidence of a compensatory mechanism involving CDC25A that ensures timely mitotic entry at different levels of cyclin A.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
-
WB
-
Biochemistry and Molecular biology
-
Cell Biology
Autocleavage of separase suppresses its premature activation by promoting binding to cyclin B1.
In Cell Reports on 29 November 2022 by Shindo, N., Kumada, K., et al.
Accurate chromosome segregation requires timely activation of separase, a protease that cleaves cohesin during the metaphase-to-anaphase transition. However, the mechanism that maintains the inactivity of separase prior to this event remains unclear. We provide evidence that separase autocleavage plays an essential role in this process. We show that the inhibition of separase autocleavage results in premature activity before the onset of anaphase, accompanied by the formation of chromosomal bridges and spindle rocking. This deregulation is attributed to the reduced binding of cyclin B1 to separase that occurs during the metaphase-to-anaphase transition. Furthermore, when separase is mutated to render the regulation by cyclin B1 irrelevant, which keeps separase in securin-binding form, the deregulation induced by autocleavage inhibition is rescued. Our results reveal a physiological role of separase autocleavage in regulating separase, which ensures faithful chromosome segregation.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
-
Homo sapiens (Human)
Mre11 exonuclease activity promotes irreversible mitotic progression under replication stress.
In Life Science Alliance on 1 June 2022 by Hashimoto, Y. & Tanaka, H.
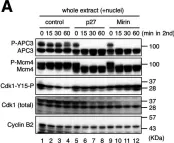
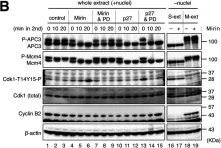
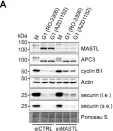
Mre11 is a versatile exo-/endonuclease involved in multiple aspects of DNA replication and repair, such as DSB end processing and checkpoint activation. We previously demonstrated that forced mitotic entry drives replisome disassembly at stalled replication forks in Xenopus egg extracts. Here, we examined the effects of various chemical inhibitors using this system and discovered a novel role of Mre11 exonuclease activity in promoting mitotic entry under replication stress. Mre11 activity was necessary for the initial progression of mitotic entry in the presence of stalled forks but unnecessary in the absence of stalled forks or after mitotic entry. In the absence of Mre11 activity, mitotic CDK was inactivated by Wee1/Myt1-dependent phosphorylation, causing mitotic exit. An inhibitor of Wee1/Myt1 or a nonphosphorylatable CDK1 mutant was able to partially bypass the requirement of Mre11 for mitotic entry. These results suggest that Mre11 exonuclease activity facilitates the processing of stalled replication forks upon mitotic entry, which attenuates the inhibitory pathways of mitotic CDK activation, leading to irreversible mitotic progression and replisome disassembly.
© 2022 Hashimoto and Tanaka.
-
WB
-
Homo sapiens (Human)
-
Cell Biology
In Life Sci Alliance on 1 June 2022 by Hashimoto, Y. & Tanaka, H.
Fig.3.A

-
WB
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 6
In Life Sci Alliance on 1 June 2022 by Hashimoto, Y. & Tanaka, H.
Fig.3.B

-
WB
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 6
In Life Sci Alliance on 1 June 2022 by Hashimoto, Y. & Tanaka, H.
Fig.4.B

-
WB
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 6
In Life Sci Alliance on 1 June 2022 by Hashimoto, Y. & Tanaka, H.
Fig.4.A

-
WB
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 6
In Biol Open on 6 March 2015 by Voets, E. & Wolthuis, R.
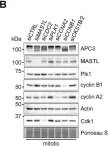
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Biol Open by CiteAb, provided under a CC-BY license
Image 1 of 6
In Biol Open on 6 March 2015 by Voets, E. & Wolthuis, R.
Fig.2.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Biol Open by CiteAb, provided under a CC-BY license
Image 1 of 6