Signal transduction pathways post-translationally regulating nucleotide metabolism remain largely unknown. Guanosine monophosphate reductase (GMPR) is a nucleotide metabolism enzyme that decreases GTP pools by converting GMP to IMP. We observed that phosphorylation of GMPR at Tyr267 is critical for its activity and found that this phosphorylation by ephrin receptor tyrosine kinase EPHA4 decreases GTP pools in cell protrusions and levels of GTP-bound RAC1. EPHs possess oncogenic and tumor-suppressor activities, although the mechanisms underlying switches between these two modes are poorly understood. We demonstrated that GMPR plays a key role in EPHA4-mediated RAC1 suppression. This supersedes GMPR-independent activation of RAC1 by EPHA4, resulting in a negative overall effect on melanoma cell invasion and tumorigenicity. Accordingly, EPHA4 levels increase during melanoma progression and inversely correlate with GMPR levels in individual melanoma tumors. Therefore, phosphorylation of GMPR at Tyr267 is a metabolic signal transduction switch controlling GTP biosynthesis and transformed phenotypes.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Product Citations: 10
In Cell Chemical Biology on 16 June 2022 by Wolff, D. W., Deng, Z., et al.
-
Genetics
In The Journal of Biological Chemistry on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
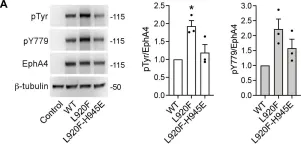
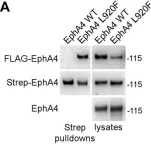
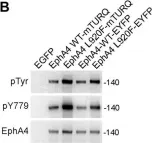
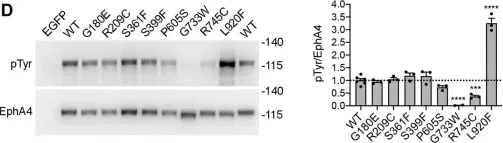
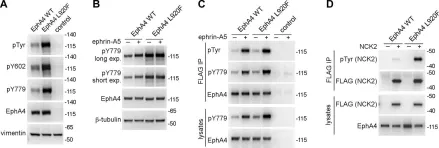
The Eph receptor tyrosine kinases and their ephrin ligands regulate many physiological and pathological processes. EphA4 plays important roles in nervous system development and adult homeostasis, while aberrant EphA4 signaling has been implicated in neurodegeneration. EphA4 may also affect cancer malignancy, but the regulation and effects of EphA4 signaling in cancer are poorly understood. A correlation between decreased patient survival and high EphA4 mRNA expression in melanoma tumors that also highly express ephrinA ligands suggests that enhanced EphA4 signaling may contribute to melanoma progression. A search for EphA4 gain-of-function mutations in melanoma uncovered a mutation of the highly conserved leucine 920 in the EphA4 sterile alpha motif (SAM) domain. We found that mutation of L920 to phenylalanine (L920F) potentiates EphA4 autophosphorylation and signaling, making it the first documented EphA4 cancer mutation that increases kinase activity. Quantitative Föster resonance energy transfer and fluorescence intensity fluctuation (FIF) analyses revealed that the L920F mutation induces a switch in EphA4 oligomer size, from a dimer to a trimer. We propose this switch in oligomer size as a novel mechanism underlying EphA4-linked tumorigenesis. Molecular dynamics simulations suggest that the L920F mutation alters EphA4 SAM domain conformation, leading to the formation of EphA4 trimers that assemble through two aberrant SAM domain interfaces. Accordingly, EphA4 wild-type and the L920F mutant are affected differently by the SAM domain and are differentially regulated by ephrin ligand stimulation. The increased EphA4 activation induced by the L920F mutation, through the novel mechanism we uncovered, supports a functional role for EphA4 in promoting pathogenesis.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cancer Research
Preprint on BioRxiv : the Preprint Server for Biology on 7 September 2020 by Banerjee, S. L., Lavoie, N., et al.
h4>SUMMARY/h4> The EPH family is the largest among receptor tyrosine kinases (RTKs) in humans. In contrast to other RTKs, EPH receptors (EPHRs) cognate ligands, ephrins, are tethered to the cell surface. This results in EPHR-ephrin signaling being mainly involved in short-range cell-cell communication events that regulate cell adhesion, migration and tissue boundary formation. Although EPHRs functions have been broadly studied, the molecular mechanisms by which they control these processes are far from being understood. To address this, we sought to identify new effector proteins acting downstream of EPHRs and determine their role in EPHR-regulated functions. To unravel EPHR-associated signaling complexes under native conditions, we applied a mass spectrometry-based approach, namely BioID proximity labeling. We obtained a composite proximity network from EPHA4, -B2, -B3 and -B4 receptors that comprises 395 proteins, most of which were not previously linked to EPH signaling. A gene ontology and pathway term analysis of the most common candidates highlighted cell polarity as a novel function associated with EPHR activity. We found that EPHA1 and EPHB4 expression is restricted to the basal and lateral membrane domains in polarized Caco-2 3D spheroidal cell cultures. We further discovered that their depletion impairs the compartmentalized distribution of polarity proteins as well as overall spheroid morphogenesis. Moreover, we examined the contribution of a number of candidates, selected from EPHR proximity networks, via loss-of-function in an EPHR-dependent cell segregation assay. We found that depletion of the signaling scaffold PAR-3 blocks cell sorting. We also delineated a signalling complex involving the C-terminal SRC kinase (CSK), whose recruitment to PAR-3 complexes is dependent on EPHR signals. Our work sheds a new light on EPHR signaling networks and describes conceptually novel the mechanisms by which EPHRs signal at the membrane to contribute to the regulation of cellular phenotypes.
In Scientific Reports on 2 October 2018 by Scilabra, S. D., Pigoni, M., et al.
The tissue inhibitor of metalloproteinases-3 (TIMP-3) is a major regulator of extracellular matrix turnover and protein shedding by inhibiting different classes of metalloproteinases, including disintegrin metalloproteinases (ADAMs). Tissue bioavailability of TIMP-3 is regulated by the endocytic receptor low-density-lipoprotein receptor-related protein-1 (LRP-1). TIMP-3 plays protective roles in disease. Thus, different approaches have been developed aiming to increase TIMP-3 bioavailability, yet overall effects of increased TIMP-3 in vivo have not been investigated. Herein, by using unbiased mass-spectrometry we demonstrate that TIMP-3-overexpression in HEK293 cells has a dual effect on shedding of transmembrane proteins and turnover of soluble proteins. Several membrane proteins showing reduced shedding are known as ADAM10 substrates, suggesting that exogenous TIMP-3 preferentially inhibits ADAM10 in HEK293 cells. Additionally identified shed membrane proteins may be novel ADAM10 substrate candidates. TIMP-3-overexpression also increased extracellular levels of several soluble proteins, including TIMP-1, MIF and SPARC. Levels of these proteins similarly increased upon LRP-1 inactivation, suggesting that TIMP-3 increases soluble protein levels by competing for their binding to LRP-1 and their subsequent internalization. In conclusion, our study reveals that increased levels of TIMP-3 induce substantial modifications in the cellular secretome and that TIMP-3-based therapies may potentially provoke undesired, dysregulated functions of ADAM10 and LRP-1.
-
WB
-
Homo sapiens (Human)
In Molecular Cell on 21 June 2018 by Dionne, U., Chartier, F. J. M., et al.
Phosphotyrosine (pTyr) signaling has evolved into a key cell-to-cell communication system. Activated receptor tyrosine kinases (RTKs) initiate several pTyr-dependent signaling networks by creating the docking sites required for the assembly of protein complexes. However, the mechanisms leading to network disassembly and its consequence on signal transduction remain essentially unknown. We show that activated RTKs terminate downstream signaling via the direct phosphorylation of an evolutionarily conserved Tyr present in most SRC homology (SH) 3 domains, which are often part of key hub proteins for RTK-dependent signaling. We demonstrate that the direct EPHA4 RTK phosphorylation of adaptor protein NCK SH3s at these sites results in the collapse of signaling networks and abrogates their function. We also reveal that this negative regulation mechanism is shared by other RTKs. Our findings uncover a conserved mechanism through which RTKs rapidly and reversibly terminate downstream signaling while remaining in a catalytically active state on the plasma membrane.
Copyright © 2018 Elsevier Inc. All rights reserved.
-
Biochemistry and Molecular biology
In J Biol Chem on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
Fig.7.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 5
In J Biol Chem on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 5
In J Biol Chem on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
Fig.4.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 5
In J Biol Chem on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
Fig.1.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 5
In J Biol Chem on 1 July 2021 by Light, T. P., Gómez-Soler, M., et al.
Fig.2.A,B,C,D

-
WB
-
Homo sapiens (Human)
Collected and cropped from J Biol Chem by CiteAb, provided under a CC-BY license
Image 1 of 5